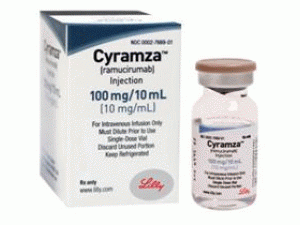
雷莫芦单抗,雷莫芦单抗注射溶液(Cyramza Infusionskonzentrat 100mg/10ml)
 药店国别:无
产地国家:瑞士
处方药:是
所属类别: 100毫克/10毫升/瓶
包装规格: 100毫克/10毫升/瓶
计价单位:瓶
生产厂家中文参考译名:无
生产厂家英文名:Eli Lilly (Suisse) S.A.
原产地英文商品名:Cyramza Infusionskonzentrat 100mg/10ml
原产地英文药品名:ramucirumab
中文参考商品译名:Cyramza注射溶液 100毫克/10毫升/瓶
中文参考药品译名:雷莫芦单抗
曾用名:无
药店国别:无
产地国家:瑞士
处方药:是
所属类别: 100毫克/10毫升/瓶
包装规格: 100毫克/10毫升/瓶
计价单位:瓶
生产厂家中文参考译名:无
生产厂家英文名:Eli Lilly (Suisse) S.A.
原产地英文商品名:Cyramza Infusionskonzentrat 100mg/10ml
原产地英文药品名:ramucirumab
中文参考商品译名:Cyramza注射溶液 100毫克/10毫升/瓶
中文参考药品译名:雷莫芦单抗
曾用名:无
简介
部份中文雷莫芦单抗处方资料(仅供参考) 英 文 名:Cyramza 商 品 名:Ramucirumab 中 文 名:雷莫芦单抗注射剂 开发厂家:礼来制药 药品介绍近日,欧盟委员会(EC)已批准单抗药物Cyramza(ramucirumab):(1)联合紫杉醇(paclitaxel),用于既往经化疗治疗的晚期胃癌或胃食管交界腺癌(GEJA)成人患者的治疗;(2)作为一种单药疗法,用于不适合紫杉醇联合疗法的晚期胃癌或胃食管交界腺癌(GEJA)成人患者的治疗。 初次批准:2014 作用机制 雷莫芦单抗是一种血管内皮生长因子受体2拮抗剂与VEGF受体2特异性结合和阻断VEGFR配体,VEGF-A,VEGF-C,和VEGF-D的结合。其结果,雷莫芦单抗抑制配体刺激的VEGF受体2的激活,因此抑制配体-诱导的增殖,和人内皮细胞的迁移。在一种体内模型中雷莫芦单抗抑制血管生成。 适应证和用途 CYRAMZA®是一个人血管内皮升长因子受体2拮抗剂适用:⑴ 作为单药或与紫杉醇联用,为晚期胃或胃-食道连接腺癌,有用或氟嘧啶[fluoropyrimidine]-或含铂化疗后疾病进展的治疗。⑵ 与多烯紫杉醇联用,为治疗转移非小细胞肺癌用或基于铂化疗后疾病进展。有EGFR或ALK基因组肿瘤畸变患者用FDA-批准的治疗对这些畸变接受CYRAMZA治疗以前应有疾病进展。 剂量和给药方法 只为静脉输注。不要静脉推注或丸注给药。胃癌⑴ CYRAMZA的推荐剂量或作为单药或与每周紫杉醇联用是8mg/kg每2周。非小细胞肺癌⑵ 在输注多烯紫杉醇前,在21天疗程在第1天静脉给予CYRAMZA 10mg/kg。 剂型和规格注射:⑴ 单剂量小瓶,100mg/10mL(10mg每mL)溶液⑵ 单剂量小瓶,500mg/50mL(10mg每mL)溶液 禁忌证:无。 警告和注意事项 ⑴ 动脉血栓栓塞事件(ATEs):在临床试验中曾报道严重,有时致命性ATEs。对严重ATEs终止CYRAMZA。 ⑵ 高血压:监视血压和治疗高血压。对严重高血压暂停CYRAMZA。对药物不能控制的高血压终止CYRAMZA。 ⑶ 输注相关反应:输注期间监视对体征和症状。 ⑷ 胃肠道穿孔:终止CYRAMZA。 ⑸ 受损伤口愈合:手术前不给CYRAMZA。 ⑹ 有肝硬变患者中临床恶化:在有Child-Pugh B或C肝硬变患者中可能发生脑病变新发作或恶化,腹水,或肝肾综合征。 ⑺ 可逆性后部白质脑病综合征:终止CYRAMZA。 不良反应 ⑴ 在单药CYRAMZA-治疗患者中观察到最常见不良反应发生率≥10%和较高安慰剂予≥2% 为高血压和腹泻。 ⑵ 在用CYRAMZA加紫杉醇治疗患者中观察到最常见不良反应发生率≥30%和比安慰剂加紫杉醇较高≥2%是疲乏,中性粒细胞减少,腹泻,和鼻出血。 ⑶ 在CYRAMZA加多烯紫杉醇治疗患者中观察到最常见不良反应发生率≥30%和比安慰剂加多烯紫杉醇较高≥2%是中性粒细胞减少,疲乏/无力,和口炎/粘膜炎症。 ⑴ 妊娠:根据它的作用机制,CYRAMZA可能致胎儿危害。 ⑵ 哺乳母亲:终止哺乳或终止CYRAMZA。英文版说明
Cyramza Infusionskonzentrat 100mg/10ml DurchstechflascheFachinformationenZusammensetzungWirkstoff: Ramucirumab.Hilfsstoffe: Histidin, Histidin-Monohydrochlorid Natriumchlorid, Glycin (E640), Polysorbat 80 (E433), Wasser für Injektionszwecke.Galenische Form und Wirkstoffmenge pro EinheitKonzentrat zur Herstellung einer Infusionslösung.Cyramza ist eine klare bis leicht opaleszente und farblose bis leicht gelbliche Lösung, pH 6,0.Jeder ml des Konzentrats zur Herstellung einer Infusionslösung enthält 10 mg Ramucirumab.Jede 10 ml Durchstechflasche enthält 100 mg Ramucirumab.Jede 50 ml Durchstechflasche enthält 500 mg Ramucirumab.Indikationen/AnwendungsmöglichkeitenMagenkarzinomCyramza ist indiziert in Kombination mit Paclitaxel für die Behandlung von erwachsenen Patienten mit fortgeschrittenem Adenokarzinom des Magens oder gastroösophagealen Übergangs mit einem Progress nach vorausgegangener Platin- und Fluoropyrimidin-haltiger Chemotherapie.Cyramza Monotherapie ist inzidiert für die Behandlung von erwachsenen Patienten mit fortgeschrittenem Adenokarzinom des Magens oder des gastroösophagealen Übergangs mit einem Progress nach vorausgegangener Platin- oder Fluoropyrimidin-haltiger Chemotherapie, wenn diese Patienten für eine Kombinationstherapie mit Paclitaxel nicht geeignet sind.Kolorektales Karzinom (mKRK)Cyramza ist in Kombination mit FOLFIRI (Irinotecan, Folinsäure und 5-Fluorouracil) indiziert zur Behandlung von erwachsenen Patienten mit metastasiertem kolorektalem Karzinom (mKRK) mit Progress während oder nach vorausgegangener Therapie mit Bevacizumab, Oxaliplatin und einem Fluoropyrimidin.Dosierung/AnwendungDosierungEine Ramucirumab-Therapie muss von Ärzten initiiert und überwacht werden, die im Bereich Onkologie erfahren sind.Adenokarzinom des Magens oder gastroösophagealen Übergangs (GEJ)Cyramza als MonotherapieDie empfohlene Dosis Ramucirumab als Monotherapie beträgt 8 mg/kg Körpergewicht alle 2 Wochen.Cyramza in Kombination mit PaclitaxelDie empfohlene Dosis von Ramucirumab beträgt 8 mg/kg an den Tagen 1 und 15 eines 28-Tage-Zyklus – vor der Paclitaxel-Infusion. Die empfohlene Dosis von Paclitaxel beträgt 80 mg/m2 als intravenöse Infusion über etwa 60 Minuten an den Tagen 1, 8 und 15 eines 28-Tage-Zyklus. Vor jeder Paclitaxel-Infusion sollte ein aktuelles Differential-Blutbild und die klinische Chemie zur Bestimmung der Leberfunktion der Patienten vorliegen. Kriterien, die vor jeder Paclitaxel-Infusion erfüllt sein müssen, sind in Tabelle 1 aufgeführt.Tabelle 1: Kriterien, die vor jeder Paclitaxel-Gabe erfüllt sein müssenKolorektales KarzinomDie empfohlene Dosis Ramucirumab beträgt 8 mg/kg alle 2 Wochen als intravenöse Infusion vor der Gabe von FOLFIRI. Vor Beginn der Chemotherapie muss das vollständige Blutbild des jeweiligen Patienten vorliegen. Bitte beachten Sie die jeweiligen Fachinformationen der Komponenten von FOLFIRI bezüglich der Anforderungen zu Prämedikation und Dosierungs-/Anwendungsempfehlungen.Dauer der TherapieEs wird empfohlen, die Behandlung bis zum Progress oder bis zum Auftreten nicht-akzeptabler Toxizität fortzusetzen.PrämedikationVor der Infusion von Ramucirumab wird eine Prämedikation mit einem Histamin-H1-Antagonisten (z.B. Diphenhydramin) empfohlen. Kam es bei einem Patienten bereits zu infusionsbedingten Reaktionen Grad 1 oder 2, muss vor allen folgenden Infusionen eine Prämedikation verabreicht werden. Nach einer zweiten infusionsbedingten Reaktion Grad 1 oder 2 soll Dexamethason (oder Äquivalent) gegeben werden. Anschliessend muss bei weiteren Infusionen eine Prämedikation mit den nachfolgenden (oder äquivalenten) Arzneimitteln erfolgen: Ein Histamin-H1-Antagonist intravenös (z.B. Diphenhydramin), Paracetamol und Dexamethason.Bitte beachten Sie die jeweiligen Fachinformationen von Paclitaxel und der Komponenten von FOLFIRI bezüglich der Prämedikationsanforderungen und zusätzlicher Informationen.DosisanpassungenInfusionsbedingte ReaktionenDie Ramucirumab Infusionsrate muss für die Dauer der Infusion und alle weiteren Infusionen um 50% reduziert werden, wenn bei einem Patient eine infusionsbedingte Reaktion Grad 1 oder 2 auftritt. Ramucirumab muss sofort und endgültig abgesetzt werden, wenn eine infusionsbedingte Reaktion Grad 3 oder 4 auftritt (siehe «Warnhinweise und Vorsichtsmassnahmen»).HypertonieDer Blutdruck des Patienten muss vor jeder Ramucirumab-Gabe überprüft und entsprechend behandelt werden, wenn es klinisch notwendig ist. Im Fall einer schweren Hypertonie muss die Ramucirumab-Therapie zeitweise gestoppt werden, bis der Blutdruck mit entsprechender Therapie wieder unter Kontrolle ist. Falls es sich um eine medizinisch signifikante Hypertonie handelt, die nicht auf verträgliche Weise mit antihypertensiver Therapie unter Kontrolle gebracht werden kann, muss die Ramucirumab-Therapie endgültig beendet werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).ProteinuriePatienten müssen hinsichtlich einer Entstehung oder Verschlechterung einer Proteinurie während der Ramucirumab-Therapie beobachtet werden. Falls der Urinstreifentest ≥2+ anzeigt, sollte der 24-Stunden-Sammelurin gemessen werden. Die Ramucirumab-Therapie muss zeitweilig abgesetzt werden, wenn die Proteinausscheidung im Urin bei ≥2 g/24 h liegt. Wenn die Proteinausscheidung im Urin wieder bei <2 g/24 h liegt, kann die Behandlung fortgesetzt werden mit einer reduzierten Dosis (6 mg/kg alle 2 Wochen). Eine zweite Dosisreduktion (auf 5 mg/kg alle 2 Wochen) wird empfohlen, wenn die Proteinausscheidung im Urin wieder auf ≥2 g/24 h ansteigt.Die Ramucirumab-Therapie muss endgültig beendet werden, wenn die Proteinausscheidung im Urin bei >3 g/24 h liegt oder im Fall eines nephrotischen Syndroms (siehe «Unerwünschte Wirkungen»).Geplante Operationen oder beeinträchtige WundheilungDie Ramucirumab-Therapie muss mind. 4 Wochen vor einer geplanten Operation vorübergehend unterbrochen werden. Bei Wundheilungskomplikationen muss die Ramucirumab-Therapie vorübergehend gestoppt werden, bis die Wunde vollständig verheilt ist (siehe «Warnhinweise und Vorsichtsmassnahmen»).Die Ramucirumab-Therapie muss endgültig beendet werden bei folgenden Ereignissen:Schwere arterielle thromboembolische Ereignisse (siehe «Warnhinweise und Vorsichtsmassnahmen»).Gastrointestinale Perforationen (siehe «Warnhinweise und Vorsichtsmassnahmen»).Schwere Blutungen: NCI CTCAE Grad 3 oder 4 Blutungen (siehe «Warnhinweise und Vorsichtsmassnahmen»).Spontane Entwicklung von Fisteln (siehe «Warnhinweise und Vorsichtsmassnahmen»).Dosisanpassungen für PaclitaxelPaclitaxel-Dosisreduktionen sollen je nach dem Grad der aufgetretenen Toxizität durchgeführt werden. Für eine hämatologische Toxizität Grad 4 gemäss NCI CTCAE oder einer nicht-hämatologischen Paclitaxel-bedingten Toxizität Grad 3 wird empfohlen, die Paclitaxel-Dosis für alle folgenden Zyklen um 10 mg/m2 zu reduzieren. Eine zweite Reduktion um 10 mg/m2 wird empfohlen, wenn die Toxizitäten persistieren oder wiederauftreten.Besondere PatientengruppenKinder und JugendlicheUnbedenklichkeit und Wirksamkeit von Cyramza bei Kindern und Jugendlichen (<18 Jahre) sind nicht erwiesen.Es liegen keine Daten vor.Es gibt in den zugelassenen Anwendungsgebieten keinen relevanten Nutzen von Cyramza bei Kindern und Jugendlichen.Ältere PatientenIn den klinischen Studien REGARD und RAINBOW gab es keinen Hinweis darauf, dass Patienten, die 65 Jahre oder älter sind, ein höheres Risiko für das Auftreten von Nebenwirkungen haben als Patienten, die jünger als 65 Jahre sind. Es werden keine Dosisreduzierungen empfohlen.Patienten mit einem Performance Status ECOG ≥2Patienten mit ECOG ≥2 waren von den pivotalen Studien ausgeschlossen. Deshalb sind Verträglichkeit und Wirksamkeit von Cyramza in dieser Patientengruppe unbekannt.Patienten mit eingeschränkter NierenfunktionFormale Studien wurden bei Patienten mit eingeschränkter Nierenfunktion nicht durchgeführt. Klinische Daten lassen vermuten, dass keine Dosisanpassung notwendig ist bei Patienten mit leichter, moderater oder schwerer Einschränkung der Nierenfunktion (siehe «Pharmakokinetik»). Es werden keine Dosisreduzierungen empfohlen.Patienten mit eingeschränkter LeberfunktionFormale Studien wurden bei Patienten mit eingeschränkter Leberfunktion nicht durchgeführt. Klinische Daten lassen vermuten, dass bei Patienten mit leichter oder moderater Einschränkung der Leberfunktion keine Dosisanpassung erforderlich ist (siehe «Pharmakokinetik»). Es gibt keine Daten zu einer Ramucirumab-Gabe bei Patienten mit schwerer Leberinsuffizienz. Es werden keine Dosisreduzierungen empfohlen.Art der AnwendungNach der Verdünnung wird Cyramza als intravenöse Infusion über etwa 60 min verabreicht. Cyramza darf nicht als intravenöse Bolusgabe verabreicht werden. Um die benötigte Infusionsdauer von etwa 60 Minuten zu erreichen, sollte eine maximale Infusionsrate von 25 mg/Minute nicht überschritten werden. Gegebenenfalls muss bei Bedarf die Infusionsdauer verlängert werden. Während der Infusion ist der Patient auf Zeichen von infusionsbedingten Reaktionen zu beobachten (siehe «Warnhinweise und Vorsichtsmassnahmen») und die Verfügbarkeit von angemessener Ausrüstung zur Reanimation muss sichergestellt sein.Hinweise zur Verdünnung des Arzneimittels vor der Anwendung, siehe «Sonstige Hinweise».KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder einen der in Zusammensetzung genannten sonstigen Bestandteile.Warnhinweise und VorsichtsmassnahmenArterielle thromboembolische EreignisseSchwere, manchmal tödlich verlaufende, arterielle thromboembolische Ereignisse (ATEs) einschliesslich Myokardinfarkt, Herzstillstand, Schlaganfall und zerebrale Ischämie wurden in klinischen Studien berichtet. Ramucirumab muss endgültig beendet werden bei Eintreten eines schweren ATE (siehe «Dosierung/Anwendung»).Gastrointestinale PerforationenRamucirumab ist eine antiangiogene Therapie und kann das Risiko für gastrointestinale Perforationen erhöhen. Es wurde über Fälle von gastrointestinalen Perforationen bei Patienten, die mit Ramucirumab behandelt wurden, berichtet. Tritt bei Patienten eine gastrointestinale Perforation auf, muss deren Behandlung mit Ramucirumab endgültig beendet werden.(siehe «Dosierung/Anwendung»).Schwere BlutungRamucirumab ist eine antiangiogene Therapie und kann das Risiko für eine schwere Blutung erhöhen. Tritt bei Patienten eine Grad 3 oder 4 Blutung auf, muss die Behandlung mit Ramucirumab endgültig abgesetzt werden (siehe «Dosierung/Anwendung»).Blutbild und Koagulationsparameter sollten, bei Patienten mit einer prädisponierenden Bedingung für Blutungen, und bei Patienten in einer Behandlung mit einem Antikoagulanz oder anderen Begleittherapien, die das Blutungsrisiko erhöhen, regelmässig überprüft werden.Schwere gastrointestinale Blutungen einschliesslich tödlicher Ereignisse wurden von Patienten mit Magenkarzinom und einer Behandlung mit Ramucirumab in Kombination mit Paclitaxel berichtet, sowie bei Patienten mit mKRK und einer Behandlung mit Ramucirumab in Kombination mit FOLFIRI.Infusionsbedingte ReaktionenInfusionsbedingte Reaktionen (infusion-related reactions, IRRs) wurden in klinischen Studien mit Ramucirumab berichtet. Die Mehrheit der Ereignisse traten auf während oder nach einer ersten oder zweiten Ramucirumab-Infusion. Patienten sollten während der Infusion auf Zeichen von Überempfindlichkeit beobachtet werden. Die Symptome beinhalten Rigor/Tremor, Rückenschmerzen/Spasmen, Brust-Schmerzen und/oder -Engegefühl, Schüttelfrost, Hitzewallungen, Dyspnoe, Giemen, Hypoxie und Parästhesie. In schweren Fällen beinhalteten die Symptome Bronchospasmen, supraventrikuläre Tachykardien und Hypotonie. Ramucirumab muss sofort und endgültig beendet werden bei Patienten mit einem IRR Grad 3 oder 4 (siehe «Dosierung/Anwendung»).HypertonieEine erhöhte Inzidenz schwerer Hypertonien wurde von Patienten unter Ramucirumab im Vergleich zu Plazebo berichtet. In den meisten Fällen wurde die Hypertonie mithilfe einer Standard-Therapie mit Antihypertensiva behandelt. Vorbestehende Hypertonie sollte unter Kontrolle gebracht werden, bevor eine Behandlung mit Ramucirumab startet. Der Blutdruck sollte überwacht werden bei Patienten, die mit Ramucirumab behandelt werden. Ramucirumab muss bei schwerer Hypertonie zeitweise gestoppt werden, bis der Blutdruck mithilfe einer Therapie unter Kontrolle gebracht ist. Wenn eine medizinisch signifikante Hypertonie mit einer antihypertensiven Therapie nicht unter Kontrolle gebracht werden kann, muss Ramucirumab endgültig beendet werden (siehe «Dosierung/Anwendung»).Beeinträchtigte WundheilungDer Einfluss von Ramucirumab wurde nicht untersucht bei Patienten mit schweren oder nicht-heilenden Wunden. Aber da Ramucirumab eine antiangiogene Therapie ist und ein Potential für einen Einfluss auf die Wundheilung haben könnte, muss die Ramucirumab-Therapie mind. 4 Wochen vor einer geplanten Operation unterbrochen werden. Wann nach dem operativen Eingriff Ramucirumab wieder gegeben werden kann, sollte anhand der klinischen Beurteilung bzgl. einer adäquaten Wundheilung entschieden werden.Falls ein Patient während der Therapie eine Wundheilungs-Komplikation erleidet, muss Ramucirumab unterbrochen werden, bis die Wunde vollständig verheilt ist (siehe «Dosierung/Anwendung»).LeberinsuffizienzRamucirumab muss mit Vorsicht angewendet werden bei Patienten mit schwerer Leberzirrhose (Child-Pugh B oder C), Zirrhose mit hepatischer Enzephalopathie, klinisch signifikanter Aszites durch Zirrhose oder einem hepatorenalen Syndrom. Bei diesen Patienten sollte Ramucirumab nur verwendet werden, wenn die individuelle Nutzen-Risiko-Abwägung positiv im Hinblick auf das Risiko eines progressiven Leberversagens einschliesslich einer hepatischen Enzephalopathie bleibt.Posteriores reversibles Leukenzephalopathie-Syndrom (PRES):Einzelfälle eines posterioren reversiblen Leukenzephalopathie-Syndroms (PRES) wurden bei Patienten berichtet, die mit Ramucirumab behandelt wurden. Das PRES kann sich manifestieren durch eine Veränderung des Geisteszustandes, epileptische Anfälle, Übelkeit, Erbrechen, Kopfschmerzen oder Sehstörungen. Die Diagnose des PRES wird mittels Magnetresonanztomografie (MRT) bestätigt. Bei Patienten, die ein PRES entwickeln, muss die Behandlung mit Ramucirumab abgebrochen werden. Die Sicherheit einer erneuten Verabreichung von Cyramza an Patienten, die zuvor ein PRES erlitten haben, ist nicht bekannt (siehe «Unerwünschte Wirkungen»).FistelnPatienten können einem erhöhten Risiko ausgesetzt sein für die Entstehung von Fisteln bei der Behandlung mit Cyramza. Ramucirumab muss beendet werden bei Patienten, die Fisteln entwickeln (siehe «Dosierung/Anwendung»).NiereninsuffizienzEs sind begrenzte Verträglichkeits-Daten verfügbar für die Therapie von Patienten mit schwerer Niereninsuffizienz (kalkulierte Creatinin-Clearance <30 ml/min.) mit Ramucirumab (siehe «Dosierung/Anwendung»).Natriumkontrollierte DiätJede 10 ml Durchstechflasche enthält ungefähr 17 mg Natrium, und jede 50 ml Durchstechflasche enthält ungefähr 85 mg Natrium. Dies sollte bei Patienten, die eine natriumkontrollierte Diät durchführen, berücksichtigt werden.InteraktionenArzneimittel-Interaktionen zwischen Ramucirumab und Paclitaxel wurden nicht beobachtet. Die Pharmakokinetik von Paclitaxel wurde durch eine Begleittherapie mit Ramucirumab nicht verändert, und die Pharmakokinetik von Ramucirumab wurde durch Paclitaxel nicht verändert. Die Pharmakokinetik von Irinotecan und seinem aktiven Metaboliten, SN-38, wurde durch eine Kombinationstherapie mit Ramucirumab nicht verändert.Schwangerschaft/StillzeitGebärfähige Frauen/Kontrazeption bei FrauenGebärfähige Frauen müssen angewiesen werden, unter Cyramza nicht schwanger zu werden und sollten über die potentielle Gefährdung für die Schwangerschaft und den Fetus aufgeklärt werden. Gebährfähige Frauen müssen effektive Massnahmen zur Kontrazeption während und bis zu 3 Monate nach Gabe der letzten Ramucirumab-Dosis treffen.SchwangerschaftEs gibt keine Daten über die Anwendung von Ramucirumab bei schwangeren Frauen. Tierstudien zur Reproduktionstoxizität mit Ramucirumab wurden nicht durchgeführt (siehe «Präklinische Daten»). Da Angiogenese kritisch ist für den Erhalt der Schwangerschaft und für die fetale Entwicklung, kann die Hemmung der Angiogenese durch Ramucirumab-Gabe zu unerwünschten Ereignissen auf die Schwangerschaft und beim Fetus führen. Cyramza sollte während der Schwangerschaft nicht angewendet werden, es sei denn, es ist klar notwendig. Wenn eine Patientin während der Therapie mit Ramucirumab schwanger wird, sollte sie über das potentielle Risiko bei Fortführung der Schwangerschaft und das Risiko für den Fetus aufgeklärt werden. Gebährfähige Frauen müssen während der Behandlung mit Cyramza eine wirksame Methode der Empfängnisverhütung anwenden.StillzeitEs ist nicht bekannt, ob Ramucirumab in die Muttermilch ausgeschieden wird. Man nimmt an, dass die Exkretion in die Milch und eine orale Aufnahme gering ist. Da ein Risiko für das Neugeborene/den Säugling nicht ausgeschlossen werden kann, sollten Frauen während der Therapie mit Cyramza das Stillen abbrechen und nach Therapieende mindestens 3 Monate nach der letzten Dosis nicht stillen.FertilitätDaten zur Wirkung von Ramucirumab auf die humane Fertilität sind nicht verfügbar. Tierstudien nach zu urteilen ist die weibliche Fertilität während der Therapie mit Ramucirumab wahrscheinlich beeinträchtigt (siehe «Präklinische Daten»).Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Studien zur Erfassung der Auswirkungen von Ramucirumab auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt. Wenn Patienten mit der Therapie zusammenhängende Symptome entwickeln, die die Fähigkeit sich zu konzentrieren und zu reagieren beeinträchtigen, wird empfohlen, dass diese so lange nicht Auto fahren oder Maschinen bedienen, bis die Symptomatik abgeklungen ist.Unerwünschte WirkungenZusammenfassung des Nebenwirkungsprofils von RamucirumabIn den pivotalen Studien wurden Ereignisse als unerwünschte Wirkungen bezeichnet, wenn sie zuvor definierte Kriterien erfüllten und als möglicherweise mit Ramucirumab zusammenhängend, klinisch relevant und medizinisch aufschlussreich angesehen wurden.Klinisch relevante unerwünschte Wirkungen (einschliesslich den schwersten unerwünschten Wirkungen Grad ≥3) assoziiert mit antiangiogener Therapie, die bei mit Ramucirumab behandelten Patienten über alle klinischen Studien (als Monotherapie oder in Kombination mit zytotoxischer Chemotherapie) hinweg beobachtet wurden, waren: gastrointestinale Perforationen, arterielle thromboembolische Ereignisse, schwere Blutungen, Hypertonie, infusionsbedingte Reaktionen und Proteinurie (siehe auch «Warnhinweise und Vorsichtsmassnahmen»).Die häufigsten unerwünschten Wirkungen, die bei Ramucirumab-behandelten Patienten beobachtet wurden, waren Neutropenie, Fatigue/Asthenie, Leukopenie, Epistaxis, Diarrhoe und Stomatitis.Tabellarische Zusammenfassung der unerwünschten WirkungenUnerwünschte Wirkungen, die von Patienten mit fortgeschrittenem Magenkarzinom oder mKRK gemeldet wurden, werden unten nach den Systemorganklassen des MedDRA-Systems, der Häufigkeit und dem Schweregrad aufgelistet. Die Häufigkeiten sind wie folgt definiert:Sehr häufig: (≥1/10)Häufig: (≥1/100 bis <1/10)Gelegentlich: (≥1/1'000 bis <1/100)Selten: (≥1/10'000 bis <1/1'000)Sehr selten: (<1/10'000)Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.MagenkarzinomRamucirumab als Monotherapie bei fortgeschrittenem MagenkarzinomDie folgende Tabelle zeigt die Häufigkeit und Schwere der Nebenwirkungen basierend auf den Ergebnissen von REGARD, einer Phase 3-Studie bei erwachsenen Patienten mit einem fortgeschrittenen Magenkarzinom – randomisiert für eine Behandlung mit Ramucirumab als Monotherapie plus Best Supportive Care (BSC) versus Plazebo plus BSC.Tabelle 2: Unerwünschte Arzneimittelreaktionen bei mit Ramucirumab behandelten Patienten in der REGARD Studie.a Bevorzugter Begriff nach MedDRA (Version 15.0)b Es gab keine Grad 5 Nebenwirkungen von Cyramza. An unerwünschten Arzneimittelwirkungen Grad 4 gab es eine (1) Hypokaliämie und eine (1) Hyponatriämie.c Siehe NCI CTCAE-Kriterien (Version 4.0) für jeden Toxizitätsgrad.d Bevorzugter Begriff nach MedDRA einschliesslich: Neutropenie und Abnahme der Neutrophilenzahle Bevorzugter Begriff nach MedDRA einschliesslich: Blut-Kaliumspiegel erniedrigt und Hypokaliämief Bevorzugter Begriff nach MedDRA einschliesslich: Angina pectoris, Herzstillstand, zerebrale Ischämie, Schlaganfall, Myokardinfarkt und myokardiale Ischämie.g Bevorzugter Begriff nach MedDRA einschliesslich: Blutdruck erhöht und Hypertonieh Bevorzugter Begriff nach MedDRA einschliesslich: Abdominalschmerzen, Unterbauchschmerzen, Oberbauchschmerzen und Leberschmerzeni Bevorzugter Begriff nach MedDRA einschliesslich: Hautausschlag, papulöser HautausschlagRamucirumab in Kombination mit Paclitaxel bei MagenkarzinomDie folgende Tabelle zeigt die Häufigkeit und Schwere der Nebenwirkungen basierend auf den Ergebnissen von RAINBOW, einer Phase 3-Studie bei erwachsenen Patienten mit einem fortgeschrittenen Magenkarzinom – randomisiert für eine Behandlung mit Ramucirumab in Kombination mit Paclitaxel oder Plazebo plus Paclitaxel.Bitte beachten Sie die Fachinformation von Paclitaxel bezüglich der bekannten unerwünschten Arzneimittelwirkungen einer Paclitaxel-Therapie.Tabelle 3: Unerwünschte Arzneimittelreaktionen bei mit Ramucirumab in Kombination mit Paclitaxel behandelten Patienten in der RAINBOW Studie.a Bevorzugter Begriff nach MedDRA einschliesslich: Mit Instrumenten zusammenhängende Sepsis, pulmonale Sepsis, Sepsis, neutropenische Sepsis, septischer Schock, Staphylokokken-Sepsis und biliäre Sepsis.b Bevorzugter Begriff nach MedDRA einschliesslich: akute Polyneuropathie, Anästhesie, axonale Neuropathie, brennendes Gefühl, Dysästhesie, Hypoästhesie, Neuralgie, Neuritis, periphere Neuropathie, Parästhesie, periphere sensorische Neuropathie, Polyneuropathie, sensorische Störungen, Verlust der Sensorik, Gefühl von Brennen der Haut und toxische Neuropathie.c einschliesslich hypertensive Kardiomyopathie.d MedDRA bevorzugter Begriff einschliesslich: anale Blutungen, blutige Diarrhoe, Magenblutungen, gastrointestinale Blutungen, Bluterbrechen, Blutstuhl, Hämorrhoiden-Blutungen, Mallory-Weiss-Syndrom, Teerstuhl, ösophageale Blutungen, rektale Blutungen und obere gastrointestinale Blutungen.e MedDRA bevorzugter Begriff einschliesslich: Angina pectoris, Herzstillstand, zerebrale Ischämie, Schlaganfall, Myokardinfarkt und myokardiale Ischämie.Kolorektales Karzinom (mKRK)Ramucirumab in Kombination mit FOLFIRIDie folgende Tabelle zeigt die Häufigkeit und Schwere der Nebenwirkungen basierend auf den Ergebnissen der RAISE-Studie, einer Phase 3-Studie bei erwachsenen Patienten mit einem metastasierten kolorektalen Karzinom (mKRK) – randomisiert für eine Behandlung mit Ramucirumab plus FOLFIRI versus Plazebo plus FOLFIRI.Tabelle 4: Unerwünschte Arzneimittelreaktionen bei mit Ramucirumab behandelten Patienten in der RAISE Studiea einschliesslich Fälle eines nephrotischen Syndroms.b MedDRA bevorzugter Begriff einschliesslich: Angina pectoris, Herzstillstand, zerebrale Ischämie, Schlaganfall, Myokardinfarkt und myokardiale Ischämie.In der RAISE-Studie bei Patienten mit mKRK und Behandlung mit Ramucirumab plus FOLFIRI war Proteinurie (1,5%) die häufigste (≥1%) Nebenwirkung, die zu einem Abbruch der Ramucriumab-Therapie geführt hat.Beschreibung ausgewählter unerwünschter WirkungenPosteriores reversibles Leukenzephalopathie-Syndrom (PRES)Im Rahmen der Phase III RAISE Studie wurde 1 Fall von PRES bei Patienten unter Ramucirumab beobachtet.Im Post-Marketing wurden Einzelfälle von PRES bei Patienten berichtet, die Ramucirumab erhalten haben (0,02%).ÜberdosierungEs gibt keine Daten zu einer Überdosierung beim Menschen. Cyramza ist in einer Phase 1-Studie untersucht worden mit bis zu 10 mg/kg alle 2 Wochen, ohne dabei eine maximal tolerierbare Dosis zu erreichen. Im Fall einer Überdosierung sollte eine unterstützende Therapie erfolgen.Eigenschaften/WirkungenATC Code: L01XC21, Antineoplastische Mittel, monoklonale AntikörperRamucirumab ist ein mittels rekombinanter DNA-Technologie aus Mäusezellen (NS0-Zellen) gewonnener, humaner, monoklonaler IgG1 Antikörper.WirkmechanismusDer vaskuläre endotheliale Wachstumsfaktorrezeptor-2 (VEGF Rezeptor-2) ist der entscheidende Vermittler der vom vaskulären endothelialen Wachstumsfaktor (VEGF) angeregten Angiogenese. Ramucirumab ist ein humaner Antikörper, der spezifisch an den VEGF Rezeptor-2 bindet, und die Bindung von VEGF-A, VEGF-C und VEGF-D blockiert. Im Ergebnis verhindert Ramucirumab die Liganden-stimulierte Aktivierung des VEGF Rezeptor-2 und seiner nachgeordneten Signalkaskaden, einschliesslich der p44/p42 Mitogen-aktivierten Proteinkinasen, und neutralisiert damit die Liganden-induzierte Proliferation und Migration der humanen Endothelzellen.Klinische Wirksamkeit und UnbedenklichkeitMagenkarzinomRAINBOWRAINBOW, eine globale, randomisierte, doppel-blinde Studie zu Cyramza plus Paclitaxel versus Plazebo plus Paclitaxel, wurde bei 665 Patienten mit lokal fortgeschrittenem und inoperablem oder metastasiertem Magenkarzinom (einschliesslich Adenokarzinom des gastroösophagealen Übergangs [GEJ]) nach einer Platin- und Fluoropyrimidin-haltigen Chemotherapie durchgeführt.Der primäre Endpunkt war das Gesamtüberleben (OS) und die sekundären Endpunkte beinhalteten progressionsfreies Überleben (PFS) und allgemeine Ansprechrate (overall response rate: ORR). Die Patienten mussten einen Progress während oder innerhalb der 4 Monate nach der letzten Dosis der First-line-Therapie gehabt haben und einen ECOG-Performance Status (PS) von 0-1 haben. Die Patienten wurden randomisiert in einem 1:1 Verhältnis für den Erhalt von Cyramza plus Paclitaxel (n = 330) oder Plazebo plus Paclitaxel (n = 335). Die Randomisierung wurde stratifiziert nach geographischer Region, Zeit bis zum Progress vom Start der First-Line-Therapie an (<6 Monate versus ≥6 Monate) und der Krankheitsmessbarkeit. Cyramza mit 8 mg/kg oder Plazebo wurden über eine intravenöse Infusion alle 2 Wochen (Tag 1 und 15) eines 28-Tage-Zyklus gegeben. Paclitaxel in einer Dosis von 80 mg/m2 wurde als intravenöse Infusion an den Tagen 1, 8 und 15 jedes 28-Tage-Zyklus gegeben.Die Mehrheit der in der Studie randomisierten Patienten (75%) erhielt vorher eine Kombination aus Platin plus Fluoropyrimidin ohne Anthrazykline. Die Übrigen (25%) erhielten zuvor Platin plus Fluropyrimidin plus Anthrazyklin. Zwei Drittel der Patienten hatten einen Progress während ihrer First-Line-Therapie (66,8%). Demographie und Charakteristika der Grunderkrankung waren grundsätzlich ausgeglichen zwischen den Armen: das mediane Alter war 61 Jahre; 71% der Patienten waren Männer; 61% waren Kaukasier, 35% Asiaten; der ECOG PS war 0 bei 39% der Patienten und 1 bei 61% der Patienten; 81% der Patienten hatten eine messbare Erkrankung und 79% hatten Magenkarzinom; 21% hatten GEJ-Adenokarzinom. Die Mehrheit der Patienten (76%) hatte einen Progress innerhalb von 6 Monaten nach Beginn der First-Line-Therapie. Für Patienten, die mit Cyramza plus Paclitaxel behandelt wurden, betrug die mediane Therapiedauer 19 Wochen, und für Patienten unter Plazebo plus Paclitaxel betrug die mediane Therapiedauer 12 Wochen.Eine ähnliche Prozentzahl an Patienten brachen die Behandlung wegen unerwünschter Ereignisse ab: 12% der Patienten, die mit Cyramza plus Paclitaxel behandelt wurden, verglichen mit 11% der Patienten, die mit Plazebo plus Paclitaxel behandelt wurden. Eine systemische Krebstherapie erhielten 47,9% der Patienten nach dem Abbruch der Therapie Cyramza plus Paclitaxel und 46,0% der Patienten , die Plazebo plus Paclitaxel erhalten hatten.Das Gesamtüberleben (OS) war statistisch signifikant verbessert bei Patienten, die Cyramza plus Paclitaxel erhalten hatten, im Vergleich zu denen, die Plazebo plus Paclitaxel erhalten hatten (HR 0,81; 95% CI: 0,68 bis 0,96; p = 0,017). Die Verlängerung des medianen Überlebens betrug 2,3 Monate für den Arm Cyramza plus Paclitaxel: 9,63 Monate im Arm Cyramza plus Paclitaxel und 7,36 Monate im Arm Plazebo plus Paclitaxel. Das progressionsfreie Überleben war bei Patienten, die Cyramza plus Paclitaxel erhielten, gegenüber Patienten unter Plazebo plus Paclitaxel statistisch signifikant verbessert (HR = 0,635, 95% CI = 0,536-0,752, p <0,0001). Die Verlängerung des medianen PFS betrug 1,5 Monate für den Arm Cyramza plus Paclitaxel: 4,4 Monate für den Arm Cyramza plus Paclitaxel und 2,9 Monate für Plazebo plus Paclitaxel. Die objektive Ansprechrate (ORR) (komplettes Ansprechen [CR] + partielles Ansprechen [PR]) von Cyramza plus Paclitaxel lag bei 27,9% und bei Plazebo plus Paclitaxel betrug sie 16,1% (Odds Rate 2,140; 95% CI: 1,499-3,160). Verbesserungen bei OS und PFS wurden konsistent beobachtet in den vorher festgelegten Subgruppen basierend auf Alter, Geschlecht, Rasse und in den meisten anderen vorher festgelegten Subgruppen.REGARDREGARD, eine multinationale, randomisierte, doppelblinde, multizentrische Studie zu Cyramza plus Best Supportive Care (BSC) versus Plazebo plus BSC wurde bei 355 Patienten mit lokal rezidiviertem und inoperabel fortgeschrittenem oder metastasiertem Magenkarzinom (einschliesslich Adenokarzinom des gastroösophagealen Übergangs [GEJ]) nach einer Platin- oder Fluoropyrimidin-haltigen Chemotherapie durchgeführt. Der primäre Endpunkt war das Gesamtüberleben (OS, Overall Survival), und Sekundärziele beinhalteten progressionsfreies Überleben (PFS) und die Rate zum progressionsfreien Überleben über 12 Wochen. Patienten mussten einen Progress während der First-Line-Behandlung gehabt haben oder einen Progress innerhalb von 4 Monaten nach der letzten Dosis der First-Line-Behandlung eines metastasierten Stadiums oder während einer adjuvanten Therapie oder innerhalb von 6 Monaten nach der letzten Dosis einer adjuvanten Therapie. Zusätzlich mussten die Patienten einen ECOG (Eastern Cooperative Oncology Group) Performance Status (PS) von 0 oder 1 haben. Um in die Studie eingeschlossen werden zu können, mussten bei den Patienten Werte von Gesamtbilirubin von ≤1,5 mg/dl bestimmt worden sein und Werte von AST und ALT ≤3-facher oberer Normalwert (ULN) oder ≤5-facher oberer Normalwert bei vorhandenen Lebermetastasen.Die Patienten wurden in einem 2:1 Verhältnis randomisiert, um entweder eine intravenöse Infusion von Cyramza 8 mg/kg (n = 238) oder Plazebo (n = 117) alle 2 Wochen zu erhalten. Die Randomisierung wurde stratifiziert nach Gewichtsverlust während der letzten 3 Monate (≥10% versus <10%), geographische Region und Sitz des Primärtumors (Magen versus gastroösophagealer Übergang).Die Patienten, die in die Studie aufgenommen wurden, hatten zuvor eine Kombinationstherapie mit Platin und Fluoropyrimidin erhalten (81%), eine Fluoropyrimidin-haltige Behandlung ohne Platin (15%) oder eine Platin-haltige Behandlung ohne Fluoropyrimidin (4%). Die 2 Behandlungsgruppen waren vergleichbar in Bezug auf Demographie und Charakteristika der Grunderkrankung: Das mediane Alter betrug 60 Jahre, 70% der Patienten waren Männer, 77% Kaukasier und 16% Asiaten; der ECOG Performance Status betrug 0 bei 28% und 1 bei 72% der Patienten; 91% der Patienten hatten eine messbare Erkrankung, 75% der Patienten hatten Magenkarzinom, 25% Adenokarzinom des GEJ. Bei der Mehrheit der Patienten (85%) war es während oder nach der First-Line Therapie zu einem Progress der Erkrankung gekommen, bei den übrigen Patienten während oder nach einer adjuvanten Therapie. Es wurden keine Patienten mit einer Leberzirrhose Child-Pugh B oder C in die REGARD Studie eingeschlossen. Die Patienten erhielten im Median 4 Zyklen (Bereich 1-34) einer Cyramza-Behandlung und 3 Zyklen (Bereich 1-30) Plazebo.11% der Cyramza-Patienten und 6% der Plazebo-Patienten brachen die Studie ab wegen unerwünschter Ereignisse. Das Gesamtüberleben war statistisch signifikant verbessert bei Patienten, die Cyramza erhalten hatten, im Vergleich zu den Patienten unter Plazebo (Hazard Rate [HR] 0,776; 95% CI: 0,603 bis 0,998; p = 0,0473), entsprechend einem um 22% verringerten Sterberisiko und einem Anstieg des medianen Überlebens auf 5,2 Monate für Cyramza von 3,8 Monate für Plazebo. Das progressionsfreie Überleben war bei Patienten, die Cyramza erhielten, gegenüber Patienten unter Plazebo statistisch signifikant verbessert (HR = 0,483, 95% CI = 0,376-0,620, p <0,0001), entsprechend einer 52% Verminderung des Progressions- oder Sterberisikos und einer Zunahme des medianen progressionsfreien Überlebens von 1,3 Monaten für Plazebo auf 2,1 Monate für Cyramza. Die Rate des progressionsfreien Überlebens nach 12 Wochen betrug 40,1% für Cyramza und 15,8% für Plazebo.Metastasiertes kolorektales KarzinomRAISEDie RAISE-Studie war eine globale, randomisierte, doppelblinde Studie zu Cyramza plus FOLFIRI versus Plazebo plus FOLFIRI und wurde bei Patienten mit einem metastasierten kolorektalen Karzinom und Progress während oder nach einer Erstlinien-Therapie mit Bevacizumab, Oxaliplatin und einem Fluoropyrimidin durchgeführt. Die Patienten mussten einen ECOG-PS von 0 oder 1 haben, und der Progress musste innerhalb von 6 Monaten nach der letzten Dosis der Erstlinien-Therapie stattgefunden haben. Insgesamt wurden 1'072 Patienten für eine Therapie mit Cyramza 8 mg/kg (n = 536) oder Plazebo (n = 536), jeweils in Kombination mit FOLFIRI im Verhältnis 1:1 randomisiert. Das FOLFIRI-Behandlungsschema bestand aus: Irinotecan 180 mg/m2 über 90 Minuten und Folinsäure 400 mg/m2 zeitgleich über 120 Minuten; gefolgt von einer Bolus-Injektion mit 5-Fluorouracil (5-FU) 400 mg/m2 über 2 bis 4 Minuten, gefolgt von 5-FU 2'400 mg/m2 als kontinuierliche Infusion über 46 bis 48 Std. In beiden Behandlungsarmen wurden die Behandlungszyklen alle 2 Wochen wiederholt. Der primäre Endpunkt war das Gesamtüberleben (OS), die sekundären Endpunkte beinhalteten das progressionsfreie Überleben (PFS).Demographische Daten und Charakteristika der Grunderkrankung in der ITT-Population waren zu Behandlungsbeginn zwischen den Behandlungsarmen ausgeglichen.Bei Patienten, die Cyramza plus FOLFIRI erhalten hatten, verbesserte sich das Gesamtüberleben (OS) statistisch signifikant im Vergleich zu Patienten, die Plazebo plus FOLFIRI erhalten hatten (HR 0,84; 95% CI: 0,73 bis 0,98; p = 0,022). Die Verlängerung des medianen Gesamtüberlebens betrug 1,6 Monate zugunsten des Behandlungsarms mit Cyramza plus FOLFIRI: 13,3 Monate im Arm Cyramza plus FOLFIRI und 11,7 Monate im Arm Plazebo plus FOLFIRI. Das progressionsfreie Überleben (PFS) war bei Patienten unter Cyramza plus FOLFIRI statistisch signifikant verbessert gegenüber Patienten unter Plazebo plus FOLFIRI (HR = 0,79, 95% CI = 0,70 bis 0,90, p = 0,0005). Die Verlängerung des medianen PFS betrug 1,2 Monate zugunsten des Arms Cyramza plus FOLFIRI: 5,7 Monate für den Arm Cyramza plus FOLFIRI und 4,5 Monate für Plazebo plus FOLFIRI.Vorab definierte Analysen des Gesamtüberlebens nach KRAS Mutationsstatus wurden durchgeführt.Vorab definierte Analysen des Gesamtüberlebens nach KRAS Mutationsstatus wurden durchgeführt. Die HR für das Gesamtüberleben betrug 0,82 (95% CI: 0,67 bis 1,0) bei Patienten mit einem KRAS-Wildtyp-Tumor und 0,89 (95% CI: 0,73 bis 1,09) bei Patienten mit einem KRAS-mutierten Tumor.ImmunogenitätPatienten aus 2 Phase III-Studien, REGARD und RAINBOW, wurden an mehreren Zeitpunkten auf Anti-Drug-Antikörper (ADAs) untersucht. Von 956 Patienten wurden Proben untersucht: 527 Ramucirumab-behandelte Patienten und 429 Kontrollpatienten. Elf (2,2%) der Ramucirumab-behandelten Patienten und zwei (0,5%) der Kontrollpatienten entwickelten ADAs. Keiner der Patienten mit ADAs entwickelte eine infusionsbedingte Reaktion (IRR). Kein Patient hatte neutralisierende Antikörper auf Ramucirumab. Die Daten sind unzureichend zur Bestimmung von Effekten der Antikörper auf die Wirksamkeit oder Verträglichkeit von Ramucirumab.PharmakokinetikBezogen auf die Dosierung von 8 mg/kg alle 2 Wochen, lag das geometrische Mittel von Ramucirumab Cmin im Serum vor Administration der vierten bzw. der siebten Dosis von Ramucirumab als Monotherapie bei Patienten mit fortgeschrittenem Magenkarzinom bei 49,5 µg/ml (Bereich 6,3-228 µg/ml) bzw. 74,4 µg/ml (Bereich 13,8-234 µg/ml).Im Serum von Patienten mit metastasiertem kolorektalem Karzinom lag das geometrische Mittel der Cmin nach einer Gabe von Ramucirumab 8 mg/kg alle 2 Wochen in Kombination mit FOLFIRI bei 46,3 μg/ml (Bereich 7,7-119 μg/ml) bzw. 65,1 μg/ml (Bereich 14,5-205 μg/ml) vor Gabe der dritten bzw. fünften Dosis.ResorptionCyramza wird als intravenöse Infusion verabreicht. Es wurden keine Studien durchgeführt mit anderen Verabreichungsarten.VerteilungBasierend auf der populations-pharmakokinetischen Analyse (PopPK) ist das mittlere Verteilungsvolumen (% Variationskoeffizient [CV %]) nach Gabe von 8 mg/kg alle 2 Wochen oder 10 mg/kg alle drei Wochen im Steady State für Ramucirumab 5,4 l (15%).MetabolismusDer Metabolismus von Ramucirumab ist nicht untersucht worden. Antikörper werden hauptsächlich durch katabole Prozesse abgebaut.EliminationBasierend auf der PopPK betrug die mittlere Clearance (CV %) von Ramucirumab nach Gabe von 8 mg/kg alle 2 Wochen oder 10 mg/kg alle drei Wochen 0,015 l/Std. (30%), und die mittlere Halbwertszeit betrug 14 Tage (20%).Kinetik spezieller PatientengruppenPatienten mit eingeschränkter NierenfunktionEs wurden keine formalen Studien durchgeführt, um die Auswirkungen einer eingeschränkten Nierenfunktion auf die Pharmakokinetik von Ramucirumab zu untersuchen.Basierend auf der PopPK war die Ramucirumab-Exposition bei Patienten mit leichter Einschränkung der Nierenfunktion (Kreatinin-Clearance [CrCl] ≥60 bis <90 ml/min, n = 687) und moderater Einschränkung der Nierenfunktion (CrCl ≥30 bis <60 ml/min, n = 244) oder schwerer Einschränkung der Nierenfunktion (CrCl 15-29 ml/min, n = 6) vergleichbar zu Patienten mit normaler Nierenfunktion (CrCl ≥90 ml/min, n = 697).Patienten mit eingeschränkter LeberfunktionEs wurden keine formalen Studien durchgeführt, um die Auswirkungen einer eingeschränkten Leberfunktion auf die Pharmakokinetik von Ramucirumab zu untersuchen.Basierend auf der PopPK war die Ramucirumab-Exposition bei Patienten mit leichter Einschränkung der Leberfunktion (Gesamtbilirubin >1,0-1,5-facher oberer Normalwert [ULN] und jegliche AST oder Gesamtbilirubin ≤1,0-facher ULN und AST >ULN, n = 525) oder moderate Einschränkung der Leberfunktion (Gesamtbilirubin >1,5-3,0-facher ULN und jegliche AST) vergleichbar zu Patienten mit normaler Leberfunktion (Gesamtbilirubin und AST ≤ ULN, n = 23). Bei Patienten mit schwerer Einschränkung der Leberfunktion (Gesamtbilirubin >3,0-facher ULN und jegliche AST) wurde Ramucirumab nicht untersucht.Präklinische DatenEs wurden keine Studien an Tieren durchgeführt, um Ramucirumab in Bezug auf sein Potential zur Karzinogenität oder Genotoxizität zu testen.Die Zielorgane, die bei wiederholter Gabe bei Cynomolgus-Affen in Toxizitätsstudien erkannt wurden, waren Niere (Glomerulonephritis), Knochen (Verdickung und abnormale endochondriale Ossifikation der Wachstumsfuge) und weibliche Reproduktionsorgane (verringertes Gewicht von Ovarien und Uterus). Ein minimaler Grad an Entzündung und/oder mononuklearer Zellinfiltration wurde in verschiedenen Organen gesehen.Reproduktionstoxizitätsstudien mit Ramucirumab wurden nicht durchgeführt. Dennoch ist anhand tierischer Modelle ein Zusammenhang zwischen Angiogenese, VEGF und VEGF Rezeptor 2 und kritischen Aspekten der weiblichen Reproduktion, embryofetalen Entwicklung und postnatalen Entwicklung erkennbar. Basierend auf dem Wirkmechanismus von Ramucirumab ist es wahrscheinlich, dass Ramucirumab bei Tieren die Angiogenese hemmen wird und damit unerwünschte Effekte auf die Fertilität (Ovulation), die Plazentareifung, Entwicklung des Fetus und die postnatale Entwicklung hat.Eine einzelne Dosis von Ramucirumab beeinträchtigte nicht die Wundheilung bei Affen bei Anwendung eines Vollhautdefekt-Modells.Sonstige HinweiseInkompatibilitätenCyramza darf nicht mit Dextrose-Lösungen zeitgleich gegeben oder gemischt werden.HaltbarkeitUngeöffnete DurchstechflascheDas Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.Nach ZubereitungEine Cyramza-Infusionslösung enthält keine antimikrobiellen Zusatzstoffe, wenn sie wie vorgeschrieben zubereitet wird.Die chemische und physikalische Stabilität von Cyramza nach Zubereitung in einer Natriumchlorid-Injektionslösung 9 mg/ml (0,9%) wurde gezeigt für: 24 Stunden bei 2 °C-8 °C bzw. für 4 Stunden bei 25 ºC.Die Infusionslösung darf nicht eingefroren oder geschüttelt werden.Besondere LagerungshinweiseAusserhalb der Reichweite von Kindern aufbewahren.Im Kühlschrank lagern (2 °C-8 °C).Nicht einfrieren.In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.Zu Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe «Haltbarkeit».Hinweise für die Handhabung und EntsorgungVerdünnung des Arzneimittels vor der Anwendung•Schütteln Sie nicht die Durchstechflasche.•Verwenden Sie bei der Zubereitung der Infusionslösung eine aseptische Technik, um die Sterilität der zubereiteten Lösung zu gewährleisten.•Jede Durchstechflasche ist nur für den einmaligen Gebrauch bestimmt. Überprüfen Sie den Inhalt der Durchstechflaschen vor der Verdünnung auf sichtbare Partikel und Verfärbungen (Cyramza 10 mg/ml Konzentrat zur Herstellung einer Infusionslösung soll klar bis leicht opaleszent und farblos bis leicht gelblich sein, ohne sichtbare Partikel). Wenn Sie sichtbare Partikel oder Verfärbungen feststellen, entsorgen Sie die Durchstechflasche.•Berechnen Sie Dosis und Volumen von Ramucirumab, wie es zur Herstellung einer Infusionslösung benötigt wird. Eine Durchstechflasche enthält entweder 100 mg oder 500 mg als 10 mg/ml Lösung Ramucirumab. Verwenden Sie nur Natriumchlorid-Injektionslösung 9 mg/ml (0,9%) als Verdünnungsmittel.Bei Nutzung von vorgefüllten Infusionsbehältnissen:Basierend auf dem berechneten Volumen Ramucirumab entnehmen Sie das entsprechende Volumen der Natriumchlorid-Injektionslösung 9 mg/ml (0,9%) aus dem vorgefüllten 250 ml Infusionsbehältnis. Überführen Sie das berechnete Volumen Ramucirumab auf aseptische Weise in das Infusionsbehältnis. Das Endvolumen in dem Behältnis sollte 250 ml betragen. Das Behältnis soll vorsichtig gewendet werden, um eine adäquate Durchmischung sicherzustellen. SCHÜTTELN SIE NICHT die Infusionslösung und FRIEREN SIE SIE NICHT EIN. Nicht mit anderen Infusionslösungen verdünnen. Nicht mit anderen elektrolythaltigen Infusionen oder Arzneimitteln über den gleichen venösen Zugang verabreichen.Bei Nutzung von nicht-vorgefüllten Infusionsbehältnissen:Überführen Sie auf aseptische Weise das berechnete Volumen an Ramucirumab in den leeren Infusionsbehälter. Geben Sie eine entsprechende Menge an Natriumchlorid-Injektionslösung 9 mg/ml (0,9%) in das Behältnis für ein Endvolumen von 250 ml. Das Behältnis soll vorsichtig gewendet werden, um eine adäquate Durchmischung sicherzustellen. SCHÜTTELN SIE NICHT die Infusionslösung und FRIEREN SIE SIE NICHT EIN. Nicht mit anderen Infusionslösungen verdünnen. Nicht mit anderen elektrolythaltigen Infusionen oder Arzneimitteln über den gleichen venösen Zugang verabreichen.Parenteral zu applizierende Arzneimittel müssen vor der Anwendung auf Partikel kontrolliert werden. Bei sichtbaren Partikeln ist die Infusionslösung zu verwerfen.Verwerfen Sie die nicht genutzte Menge an Ramucirumab, die in der Durchstechflasche verbleibt, da das Produkt keine antimikrobiellen Konservierungsmittel enthält.Applizieren Sie über eine Infusionspumpe: Für die Ramucirumab-Infusion muss ein separates Infusionsbesteck mit einem 0,22 µm-Filter mit geringer Proteinbindungskapazität genutzt werden, und das Infusionsbesteck muss mit einer Natriumchlorid-Injektionslösung 9 mg/ml (0,9%) nach Ende der Infusion gespült werden.Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.Zulassungsnummer65206 (Swissmedic).ZulassungsinhaberinEli Lilly (Suisse) S.A. Vernier/Genève用药温馨提示:当您服用此药物时,需定期接受医疗专业人士的检查,以便随时针对其药效、副作用等情况进行监测。本网站所包含的信息旨在为患者提供帮助,不能代替医学建议和治疗。
药品价格查询,专业药品查询网站,药品说明书查询,药品比价 » 雷莫芦单抗,雷莫芦单抗注射溶液(Cyramza Infusionskonzentrat 100mg/10ml)
药品价格查询,专业药品查询网站,药品说明书查询,药品比价 » 雷莫芦单抗,雷莫芦单抗注射溶液(Cyramza Infusionskonzentrat 100mg/10ml)