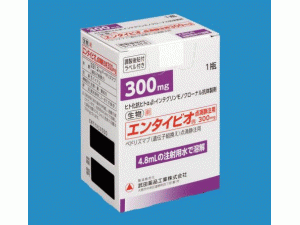
维多珠单抗冻干粉注射剂(vedolizumab)-维多珠单抗中英文说明书-Entyvio Trockensubstanz 300mg
 产地国家:瑞士
处 方 药:是
所属类别:1瓶x300毫克
包装规格:1瓶x300毫克
计价单位:瓶
生产厂家中文参考译名:
武田制药
生产厂家英文名:
Takeda Pharma AG
原产地英文商品名:
Entyvio Trockensubstanz 1x300mg
原产地英文药品名:
vedolizumab
中文参考商品译名:
Entyvio冻干粉注射剂 1瓶x300毫克
中文参考药品译名:
维多珠单抗
产地国家:瑞士
处 方 药:是
所属类别:1瓶x300毫克
包装规格:1瓶x300毫克
计价单位:瓶
生产厂家中文参考译名:
武田制药
生产厂家英文名:
Takeda Pharma AG
原产地英文商品名:
Entyvio Trockensubstanz 1x300mg
原产地英文药品名:
vedolizumab
中文参考商品译名:
Entyvio冻干粉注射剂 1瓶x300毫克
中文参考药品译名:
维多珠单抗
简介:
部份中文维多珠单抗处方资料(仅供参考) 英文名:Vedolizumab 商标名:ENTYVIO 中文名:维多珠单抗注射剂 生产商:武田制药 药品介绍 ENTYVIO(Vedolizumab) 于2014年3月21日获欧洲批准上市药品,Vedolizumab是一种人源化单克隆抗体,用于成年患者治疗对于常规治疗或对肿瘤坏死因子α(TNFα)拮抗剂应答不足、应答减弱或不耐受的中至重度活动性溃疡性结肠炎和克罗恩氏病。 作用机制 维多珠单抗是一种人源化单克隆抗体与α4β7整合素特异性结合和阻断α4β7整合素与粘膜地址素细胞[addressin cell]粘附分子-1(MAdCAM-1)的相互作用和抑制记忆T-淋巴细胞跨越内皮进入胃肠道发炎实质组织的迁移。维多珠单抗不结合至或抑制α4β1和αEβ7整合素的功能和不拮抗α4整合素与血管细胞粘附分子-1(VCAM-1)的相互作用。 优先地迁移进入胃肠道的记忆T-淋巴细胞一个离散子集的表面上表达α4β7整合素。MAdCAM-1是主要地在肠道内皮细胞上表达细胞和在T-淋巴细胞归巢[homing]至肠道淋巴组织中起至关重要作用。而α4β7整合素与MAdCAM-1的相互作用对溃疡性结肠炎和克罗恩病的标志[hallmark]的慢性炎症有重要贡献。 适应证和用途 ENTYVIO是一种整合素受体拮抗剂适用为: 成年溃疡性结肠炎(UC) ⑴ 成年有中度至严重活动性UC患者,对一种肿瘤坏死因子(TNF)阻滞剂或免疫调节剂已反应不足,失去反应,或不能耐受;或对皮质激素已反应不足,不能耐受,或显示依赖性: ⒈ 诱导和维持临床反应 ⒉ 诱导和维持临床缓解 ⒊ 改善内窥镜粘膜外观 ⒋ 实现无皮质激素缓解 成年克罗恩病[ Crohn's Disease](CD) ⑵有中度至严重活动性CD成年患者,对一种TNF阻滞剂或免疫调节剂已反应不足,失去反应,或不能耐受;或对皮质激素已反应不足,不能耐受,或显示对依赖性: ⒈ 实现临床反应 ⒉ 实现临床缓解 ⒊ 实现无皮质激素缓解 剂量和给药方法 ⑴ 在UC和CD中推荐剂量:在零,2和6周时,历时约30分钟静脉输注300 mg而其后每8周。 ⑵ 在第14周时没有显示治疗获益的患者终止ENTYVIO。 ⑶ 必须用无菌注射用水重建ENTYVIO冻干粉和给药前必须稀释在250mL无菌0.9%氯化钠。对完整重建和稀释指导见完整处方资料。 ⑷ 在重建和稀释4个小时内给予输注溶液。 ⑸ 在开始用ENTYVIO治疗前(按照当前免疫接种指导原则)给予患者所有最新免疫接种。 剂型和规格 注射用:300mg冻干维多珠单抗在一个单次使用20 mL小瓶。 英文说明书: Entyvio Trockensubstanz 300mg Durchstechflasche Fachinformationen Zusammensetzung Wirkstoff: Vedolizumab. Hilfsstoffe: L-Histidin, L-Histidin-Monohydrochlorid, L-Arginin-Hydrochlorid, Saccharose, Polysorbat 80. Galenische Form und Wirkstoffmenge pro Einheit Pulver für ein Konzentrat zur Herstellung einer Infusionslösung. Durchstechflaschen mit 300 mg Vedolizumab. 1 ml enthält 60 mg Vedolizumab nach der Rekonstitution. Indikationen/Anwendungsmöglichkeiten Colitis ulcerosa Entyvio ist indiziert für die Behandlung von Erwachsenen mit mittel- bis hochgradig aktiver Colitis ulcerosa, die auf die Standardtherapie oder einen Antagonisten von Tumornekrosefakor alpha (TNFα) nicht ausreichend oder nicht mehr ansprachen oder Unverträglichkeit zeigten. Morbus Crohn Entyvio ist indiziert für die Behandlung von Erwachsenen mit mittel- bis hochgradig aktivem Morbus Crohn, die auf die Standardtherapie oder einen Antagonisten von Tumornekrosefakor alpha (TNFα) nicht ausreichend oder nicht mehr ansprachen oder Unverträglichkeit zeigten. Dosierung/Anwendung Die Behandlung mit Entyvio ist durch spezialisiertes medizinisches Fachpersonal einzuleiten und zu überwachen, das Erfahrung in der Diagnose und Therapie von Patienten mit chronisch-entzündlichen Darmerkrankungen hat. Dosierung Erwachsene (≥18 Jahre) Das empfohlene Dosierungsschema für die Behandlung mit Entyvio beträgt 300 mg als intravenöse Infusion nach null, zwei und sechs Wochen und danach alle acht Wochen. Dieser Zeitplan gilt für Colitis ulcerosa und Morbus Crohn gleichermassen. Patienten, die eine Verminderung des Therapieansprechens zeigen, können von einer Verkürzung des Dosierungsintervalls auf 300 mg alle vier Wochen profitieren. Be Patienten mit Nicht-Ansprechen bis Woche 14 soll Entyvio abgesetzt werden. Bezüglich der Effekte der Komedikation siehe Beschreibung der klinischen Studien (Rubrik «Eigenschaften/Wirkungen»). Unter der Behandlung mit Entyvio können komedizierte Kortikosteroide unter Berücksichtigung des klinischen Ansprechens reduziert oder abgesetzt werden. Für eine Wiederaufnahme der Therapie mit Entyvio liegen keine kontrollierten Daten vor (siehe Rubrik «Klinische Wirksamkeit» und «Unerwünschte Wirkungen»). Spezielle Dosierungsanweisungen Wenn eine schwere Infusionsreaktion, insbesondere eine anaphylaktische Reaktion, auftritt, muss die Gabe von Entyvio sofort beendet und eine angemessene Behandlung eingeleitet werden (z.B. Adrenalin und Antihistaminika). Bei gering- bis mittelgradigen Infusionsreaktionen muss die Infusion verlangsamt oder unterbrochen und eine angemessene Behandlung eingeleitet werden. Sobald die gering- bis mittelgradige Infusionsreaktion abgeklungen ist, kann die Infusion wiederaufgenommen werden. Bei Patienten, die vorgängig eine gering- bis mittelgradige Infusionsreaktion hatten, sollte eine geeignete Prämedikation (z.B. mit Antihistaminika, Hydrokortison und/oder Paracetamol) vor der nächsten Infusion in Betracht gezogen werden. Ältere Patienten Daten zu Patienten >65 Jahre liegen nur beschränkt vor. Populationspharmakokinetische Analysen zeigten keinen Einfluss des Alters (siehe Rubrik «Pharmakokinetik»). Kinder und Jugendliche Die Sicherheit und Wirksamkeit von Entyvio bei Kindern und Jugendlichen im Alter von 0 bis 17 Jahren ist nicht gezeigt. Patienten mit Leber- und Niereninsuffizienz Es liegen keine Daten vor, und es können keine Dosierungsempfehlungen erfolgen. Hinweise zur Anwendung Entyvio wird vor der intravenösen Verabreichung rekonstituiert und weiter verdünnt; Anweisungen hierzu entnehmen Sie bitte der Rubrik «Sonstige Hinweise». Entyvio wird als intravenöse Infusion über 30 Minuten verabreicht. Der Patient ist während jeder Infusion kontinuierlich zu überwachen. Bei den ersten beiden Infusionen ist der Patient ausserdem nach Abschluss der Infusion noch etwa zwei Stunden lang auf Anzeichen einer akuten Überempfindlichkeitsreaktion zu beobachten. Bei allen weiteren Infusionen ist der Patient nach Abschluss der Infusion noch etwa eine Stunde lang zu beobachten. Kontraindikationen Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe. Aktive schwere Infektionen wie Tuberkulose, Sepsis, Cytomegalie, Listeriose und opportunistische Infektionen, wie z.B. progressive multifokale Leukenzephalophathie (PML) (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»). Warnhinweise und Vorsichtsmassnahmen Vor dem Beginn der Behandlung müssen alle Patienten über die Risiken der Behandlung und die Vorsichtsmassnahmen zur sicheren Anwendung von Entyvio unterrichtet werden sowie die Patientenkarte ausgehändigt bekommen. Infektionen Vedolizumab ist ein darmselektiver Integrin-Antagonist (siehe Rubrik «Wirkungsmechanismus/Pharmakodynamik»), der die Abwehrfunktion des Darmes moduliert. Ärzte sollten das möglicherweise erhöhte Risiko für opportunistische Infektionen oder Infektionen, gegen die der Darm als schützende Barriere dient, kennen (z.B. CMV-Colitis und Listeriose, siehe auch Rubrik «Unerwünschte Wirkungen»). Besondere Vorsicht ist bei der Anwendung von Entyvio bei Patienten mit einer chronischen schweren Infektion oder rezidivierenden schweren Infektionen geboten. Diese Patienten sind vor, während und nach der Therapie engmaschig auf Infektionen zu überwachen. Aktive schwere Infektionen wie Tuberkulose, Sepsis (einige mit fatalem Ausgang), Cytomegalie und Listeriose sind mit Entyvio beobachtet worden. Vor dem Beginn einer Behandlung mit Entyvio ist eine Voruntersuchung auf Tuberkulose und gegebenenfalls eine Behandlung gemäss örtlichen Richtlinien vorzunehmen. Einige Integrin-Antagonisten und einige systemische Immunsuppressiva sind mit progressiver multifokaler Leukenzephalopathie (PML) in Verbindung gebracht worden, einer seltenen und oft tödlich verlaufenden opportunistischen Infektion, die durch das John-Cunningham-Virus (JC-Virus) hervorgerufen wird. Von Entyvio ist keine systemische immunsuppressive Aktivität bekannt. In klinischen Studien mit Entyvio wurden keine Fälle von PML beobachtet, dennoch ist das Risiko einer PML nicht auszuschliessen. Bei neu auftretenden neurologischen Symptomen ist für die weiteren diagnostischen Schritte ein Neurologe beizuziehen. Falls PML vermutet wird, muss Entyvio ausgesetzt werden. Falls PML bestätigt wird, muss die Behandlung vollständig eingestellt werden. Infusions- und Überempfindlichkeitsreaktionen Jeder Patient ist während jeder Infusion kontinuierlich zu überwachen. Bei den ersten beiden Infusionen ist der Patient ausserdem nach Abschluss der Infusion noch etwa zwei Stunden lang auf Anzeichen einer akuten Überempfindlichkeitsreaktion zu beobachten. Bei allen weiteren Infusionen ist der Patient nach Abschluss der Infusion noch etwa eine Stunde lang zu beobachten (siehe Rubrik «Dosierung/Anwendung»). In klinischen Studien sind Infusions- und Überempfindlichkeitsreaktionen, einschliesslich anaphylaktoiden Reaktionen, beobachtet worden. Die meisten waren von geringem bis mittlerem Schweregrad (siehe Rubrik «Unerwünschte Wirkungen»). Immunogenität Wie bei allen Biologika besteht ein Potential für Immunogenität (siehe Rubrik «Unerwünschte Wirkungen»). Bei Patienten, die positiv für Anti-Vedolizumab-Antikörper (AVA) waren, traten gastrointestinale unerwünschte Ereignisse (wie Morbus Crohn, Colitis ulcerosa, Bauchschmerzen oder Erbrechen) häufiger auf als bei AVA-negativen Patienten. Die Anzahl der Patienten ist jedoch zu klein, um eine abschliessende Bewertung abzugeben. Impfungen Die Impfantwort bei Patienten unter einer Entyvio-Langzeittherapie ist nicht untersucht. Es wird empfohlen, bei allen Patienten den Impfschutz den aktuellen Impfempfehlungen entsprechend aufzufrischen, bevor eine Behandlung mit Entyvio begonnen wird. Totimpfstoffe können auch unter einer Entyvio-Therapie im Prinzip weiterhin angewandt werden. Nutzen und Risiken von Lebendimpfstoffen bei mit Entyvio-behandelten Patienten sind unbekannt. In einer Studie mit einer Einzeldosis Entyvio wurde bei gesunden Probanden eine reduzierte Immunantwort nach oral verabreichtem Cholera-Totimpfstoff beobachtet, wohingegen bei einer intramuskulär verabreichten Hepatitis-B-Impfung keine Abschwächung der Immunantwort beobachtet wurde. Vortherapie und Komedikation mit anderen Biologika Es liegen keine Daten aus klinischen Studien zu Entyvio bei Patienten vor, die zuvor mit Natalizumab oder Rituximab behandelt wurden. Bei diesen Patienten ist die Anwendung von Entyvio mit besonderer Vorsicht abzuwägen. Bei Patienten, die zuvor mit Natalizumab, behandelt wurden, sollte in der Regel mindestens 12 Wochen ab der letzten Dosis von Natalizumab gewartet werden, bevor eine Behandlung mit Entyvio begonnen wird. Zur gleichzeitigen Anwendung von biologischen Immunsuppressiva und Entyvio liegen keine Daten aus klinischen Studien zu Vedolizumab vor. Die Anwendung von Entyvio wird daher bei diesen Patienten nicht empfohlen. Interaktionen Entyvio ist bei Erwachsenen mit Colitis ulcerosa und Morbus Crohn bei gleichzeitiger Gabe von Kortikosteroiden, Immunmodulatoren und Aminosalicylaten untersucht worden. Populationspharmakokinetische Analysen deuten darauf hin, dass die gleichzeitige Gabe von Immunsuppressiva wie Azathioprin, 6-Mercaptopurin oder Methotrexat keinen klinisch bedeutsamen Einfluss auf die Pharmakokinetik von Vedolizumab hat. Umgekehrt ist die Interaktion von Vedolizumab mit der Pharmakokinetik von komedizierten Immunsuppressiva bisher nicht untersucht worden. Schwangerschaft/Stillzeit Schwangerschaft Es liegen nur in begrenztem Umfang Daten zur Anwendung von Entyvio bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe Rubrik «Präklinische Daten»). Entyvio sollte nicht während der Schwangerschaft angewandt werden, es sei denn der zu erwartende Nutzen überwiegt deutlich jegliche potenziellen Risiken für die Mutter und den Fötus. Frauen im gebärfähigen Alter wird dringend angeraten, während der Behandlung mit Entyvio und mindestens 18 Wochen danach eine zuverlässige Verhütungsmethode anzuwenden. Stillzeit Es ist beim Menschen nicht untersucht, ob Vedolizumab in die Muttermilch übergeht oder nach der Einnahme systemisch resorbiert wird. Da IgG-Antikörper der Mutter jedoch grundsätzlich in die Muttermilch übergehen, wird empfohlen, den Nutzen des Stillens für das Kind gegenüber dem Nutzen der Therapie für die Mutter abzuwägen und dementsprechend entweder das Stillen zu unterbrechen oder die Behandlung mit Entyvio abzusetzen bzw. darauf zu verzichten. Die vorliegenden pharmakokinetischen/toxikologischen Daten bei Tieren belegen, dass Vedolizumab in die Milch übertritt (siehe Rubrik «Präklinische Daten»). Ein Risiko für das Neugeborene/Kind kann nicht ausgeschlossen werden. Fertilität Es liegen keine Daten für die Auswirkungen von Entyvio auf die menschliche Fruchtbarkeit vor. Auswirkungen auf die männliche oder weibliche Fertilität wurden bisher nicht formell in tierexperimentellen Studien untersucht (siehe Rubrik «Präklinische Daten»). Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen Es wurden keine Studien zu den Auswirkungen auf die Fahrtüchtigkeit und das Bedienen von Maschinen durchgeführt. Entyvio kann die Fahrtüchtigkeit oder das Bedienen von Maschinen beeinträchtigen, da Schwindel als unerwünschte Wirkung aufgetreten ist (siehe Rubrik «Unerwünschte Wirkungen»). Unerwünschte Wirkungen Vedolizumab ist in drei placebokontrollierten Studien bei Patienten mit Colitis ulcerosa (GEMINI I) bzw. Morbus Crohn (GEMINI II und III) untersucht worden. In zwei kontrollierten Studien (GEMINI I und II) erhielten 620 Colitis ulcerosa bzw. 814 M. Crohn-Patienten je 300 mg Vedolizumab in Woche 0 und Woche 2 sowie danach beginnend mit Woche 6 alle acht oder alle vier Wochen für bis zu 52 Wochen, und 297 Patienten (149 Colitis ulcerosa Patienten bzw. 148 M. Crohn-Patienten) erhielten Placebo für bis zu 52 Wochen. Bei Colitis ulcerosa Patienten wurden bei 80% der mit Entyvio behandelten Patienten und bei 77% der Patienten, die Placebo erhielten, unerwünschte Ereignisse beobachtet. Im Laufe der 52 Wochen traten bei 12% der mit Entyvio behandelten Patienten schwerwiegende unerwünschte Ereignisse auf, verglichen mit 11% der Patienten, die Placebo erhielten. In den Gruppen mit achtwöchigem und vierwöchigem Dosierungsintervall waren vergleichbare Raten unerwünschter Ereignisse zu verzeichnen. Der Anteil der Patienten, die die Behandlung wegen unerwünschten Ereignissen abbrachen, betrug unter Entyvio 6% und in der Placebogruppe 11%. Die häufigsten unerwünschten Wirkungen (≥5% und mit höherer Inzidenz als unter Placebo) bei Colitis ulcerosa Patienten waren Kopfschmerzen, Nasopharyngitis, Arthralgie, Infektionen der oberen Atemwege, Husten, Bauchschmerzen, Müdigkeit, und Influenza. Bei Patienten mit M. Crohn waren bei 87% der mit Entyvio behandelten Patienten und bei 80% der Patienten, die Placebo erhielten, unerwünschte Ereignisse beobachtet worden. Im Laufe der 52 Wochen traten bei 24% der mit Entyvio behandelten Patienten schwerwiegende unerwünschte Ereignisse auf, verglichen mit 16% der Patienten, die Placebo erhielten. In den Gruppen mit achtwöchigem und vierwöchigem Dosierungsintervall waren vergleichbare Raten unerwünschter Ereignisse zu verzeichnen. Der Anteil der Patienten, die die Behandlung wegen unerwünschter Ereignisse abbrachen, betrug unter Entyvio 11% und in der Placebogruppe 9%. Die häufigsten unerwünschten Wirkungen (≥5% und mit höherer Inzidenz als unter Placebo) bei Patienten mit M. Crohn waren Arthralgie, Fieber, Nasopharyngitis, Übelkeit, Müdigkeit, und Rückenschmerzen. Alle Patienten (n = 1822; 704 Colitis ulcerosa Patienten und 1118 M. Crohn-Patienten), die zuvor in Vedolizumab-Studien der Phase II oder III aufgenommen worden waren, hatten die Möglichkeit, anschliessend an einer fortlaufenden Open-Label-Studie teilzunehmen, in der sie alle vier Wochen mit Entyvio 300 mg behandelt wurden. Unerwünschte Wirkungen Die folgende Auflistung der unerwünschten Wirkungen basiert auf den Erfahrungen aus den klinischen Studien und wird nach Organsystemen aufgelistet. Innerhalb der Systemorganklassen sind die unerwünschten Wirkungen nach ihrer Häufigkeit in folgende Kategorien eingeteilt: sehr häufig (≥1/10); häufig (≥1/100<1/10) und gelegentlich (≥1/1.000<1/100). Innerhalb jeder Häufigkeitsgruppe werden die unerwünschten Wirkungen nach abnehmendem Schweregrad angegeben. Infektionen und Infestationen Sehr häufig: Nasopharyngitis (11%). Häufig: Bronchitis, Gastroenteritis, Infektionen der oberen Atemwege, Influenza, Sinusitis, Pharyngitis. Gelegentlich: Atemwegsinfektionen, Vulvovaginalkandidose, Mundsoor. Störungen des Nervensystems Sehr häufig: Kopfschmerzen (12%). Häufig: Parästhesie. Gefässe Häufig: Hypertonie. Atmungsorgane (respiratorische, thorakale und mediastinale Funktionsstörungen) Häufig: Oropharyngeale Schmerzen, verstopfte Nase, Husten. Gastrointestinale Störungen Häufig: Analer Abszess, Analfissur, Übelkeit, Dyspepsie, Verstopfung, aufgeblähter Bauch, Blähungen, Hämorrhoiden. Funktionsstörungen der Haut und des Unterhautzellgewebes Häufig: Hautausschlag, Pruritus, Ekzem, Erythem, Nachtschweiss, Akne. Gelegentlich: Follikulitis. Muskelskelettsystem (Funktionsstörungen des Bewegungsapparates, des Bindegewebes und der Knochen) Sehr häufig: Arthralgie (12%). Häufig: Muskelkrämpfe, Rückenschmerzen, Muskelschwäche, Müdigkeit. Allgemeine Störungen und Reaktionen an der Applikationsstelle Häufig: Fieber. Gelegentlich: Reaktion an der Infusionsstelle (einschliesslich: Schmerzen und Reizungen an der Einstichstelle), Infusionsreaktionen, Schüttelfrost, Kältegefühl. Beschreibung ausgewählter unerwünschter Wirkungen Infektionen In den kontrollierten Studien GEMINI I und II betrug die Infektionsrate 0.85 pro Patienten-Jahr bei den mit Entyvio behandelten Patienten und 0.70 pro Patienten-Jahr bei den Patienten, die Placebo erhielten. Bei den Infektionskrankheiten handelte es sich vorwiegend um Nasopharyngitis, Infektionen der oberen Atemwege, Sinusitis und Harnwegsinfektionen. Die meisten Patienten wurden nach dem Abklingen der Infektion mit Entyvio weiterbehandelt. Die Rate schwerwiegender Infektionen betrug 0.07 pro Patienten-Jahr bei den mit Entyvio behandelten Patienten und 0.06 pro Patienten-Jahr bei den Patienten, die Placebo erhielten. Es wurde keine signifikante Zunahme der Häufigkeit schwerwiegender Infektionen im Zeitverlauf festgestellt. In kontrollierten und offenen Studien zu Entyvio bei Erwachsenen wurden Fälle von schwerwiegenden Infektionen gemeldet, darunter Tuberkulose, Sepsis (in einigen Fällen mit Todesfolge), Salmonellen-Sepsis, Listerien-Meningitis und Cytomegalievirus-Colitis. Maligne Erkrankungen Das Krebsrisiko ist bei Patienten mit Colitis ulcerosa und Morbus Crohn erhöht. Immunmodulatorische Arzneimittel können das Risiko für maligne Erkrankungen erhöhen. In den Studien GEMINI I und II wurden maligne Erkrankungen (ohne Dysplasie und Basalzellkarzinom) bei 6 von 1434 (0.4%) mit Entyvio behandelten Patienten berichtet; dies schliesst Darmkrebs (n = 2), Übergangsepithelkarzinom (n = 1), Brustkrebs (n = 1), Appendixkarzinoid (n = 1) und Plattenepithelkarzinom (n = 1) mit ein. Maligne Erkrankungen wurden bei einem Patienten von 297 (0.3%) mit Placebo behandelten Patienten gemeldet (Plattenepithelkarzinom). Colondysplasien kamen bei vier Patienten vor. Insgesamt deuten die Ergebnisse des bisherigen klinischen Studienprogramms nicht auf ein erhöhtes Krebsrisiko unter Entyvio hin; allerdings war die Zahl der malignen Erkrankungen gering, und die Daten zu langfristiger Exposition waren begrenzt. Langzeituntersuchungen zur Sicherheit dauern noch an. Infusionsreaktionen In den kontrollierten Studien GEMINI I und II traten bei 4% der mit Entyvio behandelten Patienten und 3% der Placebo-Patienten unerwünschte Ereignisse auf, die durch den Prüfarzt als Infusionsreaktionen eingestuft wurden(siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen») und sich typischerweise in Form von Übelkeit, Kopfschmerzen, Pruritus, Schwindel, Müdigkeit, Fieber, Urtikaria und Erbrechen äusserten. Die Mehrheit der Infusionsreaktionen trat innerhalb der ersten zwei Stunden auf, war von geringer oder mittlerer Intensität und <1% führten zum Abbruch der Studienbehandlung. Eine schwerwiegende Infusionsreaktion wurde bei einem Morbus-Crohn-Patienten während seiner zweiten Infusion beobachtet (mit den Symptomen Dyspnoe, Bronchospasmus, Urtikaria, Hautröte, Exanthem und Erhöhung des Blutdrucks und der Herzfrequenz); die Reaktion wurde erfolgreich beherrscht, indem die Infusion beendet und der Patient mit Antihistaminika und intravenösem Hydrokortison behandelt wurde. Bei Patienten, die in Woche 0 und 2 Entyvio und anschliessend Placebo erhielten, wurde bei Wiederaufnahme der Therapie keine erhöhte Infusionsreaktionsrate beobachtet. Immunogenität In den kontrollierten Studien GEMINI I und II zeigte Vedolizumab eine Immunogenitätsrate von 4% (56 der 1434 Patienten, die kontinuierlich mit Vedolizumab behandelt wurden, waren während der Behandlung einmal positiv für anti-Vedolizumab-Antikörper). 9 dieser 56 Patienten waren persistent positiv (anti-Vedolizumab-Antikörper bei zwei oder mehr Studienterminen), und 33 Patienten entwickelten neutralisierende anti-Vedolizumab-Antikörper. Die Häufigkeit, mit der Antikörper gegen Vedolizumab nach der letzten Dosis der Studienmedikation (18 Wochen und somit etwa fünf Halbwertszeiten nach der letzten Dosis) bei den Teilnehmern von GEMINI I und II nachgewiesen wurden, betrug etwa 10%. In den kontrollierten Studien GEMINI I und II waren 5% (3 von 61) der Patienten, bei denen ein vom Prüfarzt als Infusionsreaktion eingestuftes unerwünschtes Ereignis aufgetreten war, persistent positiv für anti-Vedolizumab-Antikörper. Von den 4 Patienten mit Infusionsreaktionen, die zum Studienabbruch führten, waren 2 Antikörper-positiv. Überdosierung Es wurde keine dosislimitierende Toxizität in klinischen Studien beobachtet. Dosierungen von bis zu 10 mg/kg (entspricht etwa dem 2.5-fachen der empfohlenen Dosis) wurden im Rahmen klinischer Studien verabreicht. In diesen klinischen Studien wurden keine Fälle von Überdosierung beobachtet. Wirkungsmechanismus/Pharmakodynamik Vedolizumab ist ein selektiv im Darm wirkender immunmodulatorischer humanisierter monoklonaler Antikörper, der an das α4β7-Integrin auf pathogenen Homing-Lymphozyten im Darm bindet und selektiv die Adhäsion dieser Zellen an das mukosale Addressin-Zelladhäsionsmolekül 1 (MAdCAM-1) hemmt, nicht aber an das vaskuläre Zelladhäsionsmolekül 1 (VCAM-1). Vedolizumab bindet nicht an α4β1- und αEβ7-Integrine und inhibiert nicht deren Funktion. In therapeutischen Dosen wird das Zielmolekül abgesättigt. α4β7-Integrin wird auf der Oberfläche einer diskreten Teilmenge von Gedächtnis-T-Lymphozyten exprimiert, die vorzugsweise in den Gastrointestinaltrakt einwandern. MAdCAM-1 wird hauptsächlich von den Darm-Endothelzellen exprimiert und spielt eine entscheidende Rolle bei der Einwanderung von T-Lymphozyten in Lymphgewebe im Darm. Die Interaktion von αβ7 mit MAdCAM-1 wird als wichtiger Beitrag für die chronische Entzündung, die das Kennzeichen für Colitis ulcerosa und M. Crohn ist, betrachtet. Bei Patienten mit Colitis ulcerosa bewirkte Vedolizumab eine Verbesserung der intestinalen Entzündung, die auch histopathologisch und endoskopisch nachweisbar war. Ausserdem hemmt Vedolizumab die Immunantwort der Darmmukosa auf eine gastrointestinale Antigen-Provokation, nicht aber auf eine intramuskuläre Antigen-Provokation bei gesunden menschlichen Probanden. Weder bei gesunden Probanden noch bei Patienten mit Colitis ulcerosa oder Morbus Crohn bewirkt Vedolizumab einen Anstieg der Neutrophilen, Basophilen, Eosinophilen, B-Helferzellen, zytotoxischen T-Lymphozyten, der gesamten Gedächtnis-T-Helferzellen, Monozyten oder natürlichen Killerzellen im peripheren Blut. Vedolizumab hatte keinen Einfluss auf die Wanderung von CD4+ und CD8+ ins ZNS, wie das gleichbleibende Verhältnis von CD4+ zu CD8+ im Liquor gesunder Probanden vor und nach Vedolizumab-Gabe belegt. Der Einfluss von Vedolizumab auf das Migrationsverhalten von Entzündungszellen bei entzündlichen und/oder infektiösen Prozessen im ZNS wurde am Menschen nicht untersucht. Klinische Wirksamkeit Colitis ulcerosa Die Sicherheit und Wirksamkeit von Entyvio in der Behandlung erwachsener Patienten mit mittel- bis hochgradig aktiver Colitis ulcerosa (Mayo-Score 6–12 mit Endoskopie-Subscore ≥2) wurde in einer randomisierten, doppelblinden, placebokontrollierten Studie zur Beurteilung von Wirksamkeitsendpunkten nach Woche 6 und Woche 52 (GEMINI I) nachgewiesen. Aufgenommen wurden Patienten, bei denen zuvor mindestens eine konventionelle Therapie (z.B. Kortikosteroide, Immunmodulatoren) und/oder ein oder mehrere TNFα-Antagonisten versagt hatten (einschliesslich primärer Non-Responder, sekundärer Non-Responder und Patienten mit Unverträglichkeit). Die gleichzeitige Verabreichung von oralen Aminosalicylaten, Kortikosteroiden und/oder Immunmodulatoren in stabiler Dosierung war zulässig. Zu Studienbeginn betrug der mittlere Mayo-Score 8.6; bei 37% der Patienten lag eine Pankolitis vor, und bei 41% war zuvor eine Behandlung mit TNFα-Antagonisten erfolglos. Patienten aus Gemini I konnten in die laufende offene Extensionsstudie GEMINI Open-Label-Extension (GEMINI OLE) mit Entyvio 300 mg alle 4 Wochen überführt werden. Induktion: Zur Auswertung der Endpunkte nach Woche 6 wurden 374 Patienten mittels doppelt verblindeter Randomisierung (3:2) der Behandlung mit Entyvio 300 mg oder Placebo in Woche 0 und Woche 2 zugeteilt. Der primäre Endpunkt war der Anteil der Patienten mit klinischem Ansprechen (definiert als Rückgang des Mayo-Gesamtscores um ≥3 Punkte und ≥30% gegenüber dem Ausgangswert bei gleichzeitigem Rückgang des Subscores für rektale Blutungen um ≥1 Punkt oder des absoluten Subscores für rektale Blutungen auf ≤1 Punkt) in Woche 6. Tabelle 1 zeigt die Ergebnisse der primären und sekundären Endpunkte der Studie. Klinische Remission: Mayo-Gesamtscore ≤2 Punkte und kein Subscore >1 Punkt Abheilung der Mukosa: Mayo-Subscore für den endoskopischen Befund ≤1 Punkt * Die Placebogruppe umfasst diejenigen Teilnehmer, die in Woche 0 und Woche 2 Entyvio und von Woche 6 bis Woche 52 nach Randomisierung Placebo erhielten. Anhaltendes klinisches Ansprechen: Klinisches Ansprechen nach Woche 6 und Woche 52 Anhaltende klinische Remission: Klinische Remission nach Woche 6 und Woche 52 Kortikosteroid-freie klinische Remission: Patienten, die zu Studienbeginn orale Kortikosteroide einnahmen und diese ab Woche 6 abgesetzt hatten und sich nach Woche 52 in klinischer Remission befanden. Ungefähr 40% der Gesamtpopulation von GEMINI I hatte eine erfolglose Vorbehandlung mit TNFα-Antagonisten. Die günstige Auswirkung von Entyvio bezüglich klinischem Ansprechen, Remission und Abheilung der Darmschleimhaut wurde sowohl bei TNFα-Antagonisten-naiven Patienten als auch bei Patienten mit einer erfolglosen TNFα-Antagonisten-Vortherapie gegenüber Placebo beobachtet. Gemäss explorativen Subgruppenanalysen in Woche 6 von Patienten mit Immunmodulatoren (Azathioprin, 6-Mercaptopurin) als Begleitmedikation zu Studienbeginn (Woche 0) erreichten 53% der mit Entyvio behandelten Patienten und 34% der Patienten unter Placebo ein klinisches Ansprechen (95% Konfidenzintervall für die Differenz der klinischen Ansprechrate zwischen der Entyvio- und Placebogruppe 1% -37%). In Woche 6, erreichten von den Patienten mit Kortikosteroiden als Begleitmedikation zu Studienbeginn (Woche 0), 49% unter Entyvio und 30% unter Placebo klinisches Ansprechen (95% Konfidenzintervall für die Differenz der klinischen Ansprechrate zwischen der Entyvio- und Placebogruppe 6% -33%). Erhaltung: Während der Induktionsphase in GEMINI I erhielten zwei Patientenkohorten in Woche 0 und Woche 2 Entyvio: Die Patienten in Kohorte 1 (n = 374) erhielten nach doppelt verblindeter Randomisierung entweder 300 mg Vedolizumab oder Placebo; die Patienten in Kohorte 2 (n = 521) wurden offen mit Entyvio 300 mg behandelt. Zur Beurteilung der Wirksamkeit nach Woche 52 wurden diejenigen 373/746 (50%) Patienten aus den gepoolten Kohorte 1 und 2, die unter Entyvio in Woche 6 ein klinisches Ansprechen (Rückgang des Mayo-Gesamtscores um ≥3 Punkte und ≥30% des Ausgangswerts bei gleichzeitigem Rückgang des Subscores für rektale Blutungen um ≥1 Punkt oder des absoluten Rektalblutungs-Subscores auf ≤1 Punkt) zeigten, durch doppelt verblindete Randomisierung im Verhältnis 1:1:1 einem der folgenden Behandlungsschemata ab Woche 6 zugeteilt: Entyvio 300 mg alle acht Wochen, Entyvio 300 mg alle vier Wochen oder Placebo alle vier Wochen. Placebo-Patienten aus der Induktionsphase wurden bis Woche 52 mit Placebo weitergeführt. Ab Woche 6 wurde bei den Patienten, die klinisches Ansprechen gezeigt hatten und Kortikosteroide erhielten, mit dem Ausschleichen der Kortikosteroide begonnen. Der primäre Endpunkt war der Anteil der Patienten in klinischer Remission nach Woche 52. Tabelle 2 zeigt eine Zusammenfassung der Ergebnisse der primären und sekundären Endpunkte der Erhaltungsphase. Explorative Analysen lieferten zusätzliche Daten zu wichtigen Teilpopulationen der Studie. Bei etwa einem Drittel der Patienten hatte zuvor eine Behandlung mit TNFα-Antagonisten fehlgeschlagen. Von diesen Patienten zeigten 37% der mit Entyvio alle acht Wochen und 35% der mit Entyvio alle vier Wochen behandelten Patienten sowie 5% der Patienten unter Placebo nach Woche 52 eine klinische Remission (jeweilige 95%-Konfidenzintervall für die Differenz zur klinischen Remissionsrate der Placebogruppe 10-51% bzw. 7-49%). Eine Kortikosteroid-freie Remission und Abheilung der Mukosa war in 23%, 32% und 4% bzw. in 42%, 48% und 8% der Patienten, bei denen eine Vorbehandlung mit TNFα Antagonisten versagt hatte, nach Verabreichung von Entyvio alle 8 Wochen, Entyvio alle 4 Wochen und Placebo zu verzeichnen. In explorativen Subgruppenanalysen von den Patienten mit komedizierten Immunmodulatoren (Azathioprin, 6-Mercaptopurin) zu Studienbeginn (Woche 0) zeigten in Woche 52 44% unter Entyvio alle acht Wochen und 47% unter Entyvio alle vier Wochen sowie 20% unter Placebo eine klinische Remission (jeweilige 95-%-Konfidenzintervalle für die Differenz zur klinischen Remissionsrate der Placebogruppe 6–43% bzw. 9–45%). Von den Patienten mit komedizierten Kortikosteroiden zu Studienbeginn (Woche 0) zeigten in Woche 52 36% unter Entyvio alle acht Wochen und 51% unter Entyvio alle vier Wochen sowie 15% unter Placebo eine klinische Remission (jeweilige 95%-Konfidenzintervalle für die Differenz zur klinischen Remissionsrate der Placebogruppe 7–34% bzw. 21–50%). Es erfolgte auch eine Beurteilung der gesundheitsbezogenen Lebensqualität (HRQoL) anhand des krankheitsspezifischen Fragebogens IBDQ (Inflammatory Bowel Disease Questionnaire) sowie der allgemeinen Fragebögen SF-36 und EQ-5D. Bei den Entyvio-Gruppen wurden nach Woche 6 und Woche 52 klinisch bedeutsame Verbesserungen festgestellt, die signifikant stärker ausgeprägt waren als in der Placebogruppe. Dies betraf den EQ-5D- und EQ-5D-VAS-Score, alle Subskalen des IBDQ (Darm, systemisch, emotional, sozial) und alle Subskalen des SF-36 einschliesslich Physical Component Summary (PCS) und Mental Component Summary (MCS). Die Daten der offenen Extensionsstudie mit Entyvio alle 4 Wochen sind mit einem anhaltenden Nutzen hinsichtlich Mayo-Subscores, klinischer Remission und klinischem Ansprechen bis zu 124 Wochen vereinbar. Die offene Extensionsstudie schloss 675 Patienten mit colitis ulcerosa aus GEMINI I ein. Verzögertes Ansprechen: Patienten, die nach Woche 6 unter Entyvio bzw. Placebo kein Ansprechen zeigten, verblieben in der Studie und erhielten weiterhin alle vier Wochen Entyvio bzw. Placebo. Klinisches Ansprechen laut Mayo-Subscores lag nach Woche 10 und Woche 14 unter Entyvio bei 32% bzw. 39% vor gegenüber 15% bzw. 21% unter Placebo. Wiederaufnahme der Therapie: Patienten, die unter Entyvio in Woche 6 angesprochen hatten und dann nach Placebo randomisiert wurden und im weiteren Verlauf einen Verlust des Ansprechens erfuhren, durften in die laufende offene Extensionsstudie GEMINI OLE wechseln, wo sie Entyvio alle vier Wochen erhielten. Bei 45% dieser Patienten wurde eine klinische Remission nach Woche 28 erreicht, bei 36% nach Woche 52. Dosiseskalation: Patienten, die nach anfänglichem Ansprechen mit Entyvio alle acht Wochen einen Verlust des Ansprechens erfuhren, konnten in die offene Extensionsstudie GEMINI OLE wechseln, wo sie Entyvio alle vier Wochen erhielten. Bei 25% dieser Patienten war eine klinische Remission in Woche 28 und Woche 52 zu verzeichnen. Hospitalisationen, operative Eingriffe und Kolektomien: In GEMINI I betrug bis Woche 52 der Anteil der CU-bedingten Krankenhausaufenthalte, Kolektomien und CU-bedingten Operationen unter TNFα-Antagonisten-naiven Patienten und Patienten nach erfolgloser TNFα-Antagonisten-Vortherapie mit Entyvio alle 8 Wochen, alle vier Wochen oder Placebo jeweils 2.8%, 4.1% und 8.9% bzw. 4.7%, 5.0% und 7.9%. In der laufenden offenen Extensionsstudie GEMINI OLE, welche allen Entyvio-Patienten ungeachtet ihres Status in der Studie GEMINI I offen stand, betrug die Kaplan-Meier-Schätzung für den Anteil CU-bedingter Krankenhausaufenthalte, Kolektomien und CU-bedingter Operationen unter TNFα-Antagonisten-naiven Patienten und Patienten mit erfolgloser TNFα-Antagonisten-Vortherapie 5.0% und 15.0% nach 1 Jahr sowie 8.2% und 17.8% nach 2 Jahren. Unter den total 657 aus GEMINI I aufgenommenen Patienten hatte bei 39% eine Therapie mit einem TNFα-Antagonisten zuvor versagt und 40% von denen, die in der Induktionsphase Entyvio erhielten, hatten bis Woche 6 kein klinisches Ansprechen gezeigt. Morbus Crohn Die Sicherheit und Wirksamkeit von Entyvio in der Behandlung erwachsener Patienten mit mittel- bis hochgradig aktivem Morbus Crohn (Morbus-Crohn-Aktivitätsindex [CDAI] von 220–450) wurden in zwei Studien untersucht, GEMINI II und III. Aufgenommen wurden Patienten, bei denen zuvor mindestens eine konventionelle Therapie (z.B. Kortikosteroide, Immunmodulatoren) und/oder TNFα-Antagonisten versagt haben (einschliesslich primärer Non-Responder). Die gleichzeitige Verabreichung von oralen Kortikosteroiden, Immunmodulatoren und/oder Antibiotika in stabiler Dosierung war zulässig. Zu Studienbeginn betrug der mittlere CDAI-Score über beide Studien hinweg 319; 14% der Patienten hatten eine Fistel mit Drainage, und 63% hatten eine erfolglose Vorbehandlung mit TNFα-Antagonisten hinter sich. Patienten aus GEMINI II konnten in die offene Extensionsstudie GEMINI OLE mit Entyvio 300 mg alle 4 Wochen überführt werden. Induktion: Die Studie GEMINI II war eine randomisierte, doppelblinde, placebokontrollierte Studie zur Beurteilung von Wirksamkeitsendpunkten nach Woche 6 und Woche 52. Bei knapp 50% der gesamten Population der GEMINI-II-Studie hatte eine vorausgehende Behandlung mit TNFα-Antagonisten versagt. Die Patienten (n = 368) wurden mittels doppelt verblindeter Randomisierung (3:2) der Behandlung mit Entyvio 300 mg oder Placebo in Woche 0 und Woche 2 zugeteilt. Die beiden primären Endpunkte waren der Anteil der Patienten in klinischer Remission (definiert als CDAI-Score ≤150 Punkte) nach Woche 6 sowie der Anteil der Patienten mit verbessertem klinischem Ansprechen («enhanced clinical response» definiert als Rückgang des CDAI um ≥100 Punkte gegenüber Studienbeginn) nach Woche 6. Primär- und Sekundärendpunkte sind in Tabelle 3 zusammengefasst. * Die Placebogruppe umfasst diejenigen Teilnehmer, die in Woche 0 und Woche 2 Entyvio und von Woche 6 bis Woche 52 nach Randomisierung Placebo erhielten. Kortikosteroid-freie klinische Remission: Patienten, die zu Studienbeginn orale Kortikosteroide einnahmen und diese ab Woche 6 abgesetzt hatten und sich nach Woche 52 in klinischer Remission befanden. Die Patientenzahlen betrugen hierbei n = 82 Placebo, n = 82 Entyvio alle acht Wochen und n = 80 Entyvio alle vier Wochen. Anhaltende klinische Remission: Klinische Remission bei ≥80% der Studientermine einschliesslich des Abschlusstermins (Woche 52) Fast 50% der Population in der Induktionsphase der GEMINI-II-Studie hatte eine vorausgehende erfolglose Behandlung mit einem TNFα Antagonisten. Unter diesen Patienten, erzielten 10% der Patienten, die Entyvio erhielten, und 4% der Patienten unter Placebo eine klinische Remission in Woche 6 (95% Konfidenzintervall für die Differenz zu Placebo -9% -21%). Unter den Patienten mit Immunmodulatoren als Begleitmedikation zu Studienbeginn (Woche 0) erzielten 13% der Patienten, die Entyvio erhielten, und 8% der Patienten unter Placebo eine klinische Remission in Woche 6 (95% Konfidenzintervall für die Differenz zu Placebo -12% -23%). Unter den Patienten mit Kortikosteroiden als Begleitmedikation zu Studienbeginn (Woche 0) erzielten 20% der Patienten, die Entyvio erhielten und 6% der Patienten unter Placebo eine klinische Remission in Woche 6 (95% Konfidenzintervall für die Differenz zu Placebo -1% -29%) gegenüber 10% unter Entyvio und 8% unter Placebo bei Patienten ohne Kortikosteroidkomedikation. Erhaltung: Während der Induktionsphase in GEMINI II erhielten zwei Patientenkohorten in Woche 0 und Woche 2 Entyvio: Die Patienten in Kohorte 1 (n = 368) erhielten nach doppelt verblindeter Randomisierung entweder 300 mg Entyvio oder Placebo; die Patienten in Kohorte 2 (n = 747) wurden offen mit Entyvio 300 mg behandelt. Zur Beurteilung der Wirksamkeit nach Woche 52 wurden diejenigen 461/967 (48%) Patienten aus den gepoolten Kohorten 1 und 2, die unter Entyvio in Woche 6 ein klinisches Ansprechen zeigten (definiert als Rückgang des CDAI-Score um ≥70 Punkte gegenüber Studienbeginn), durch doppelt verblindete Randomisierung im Verhältnis 1:1:1 einem der folgenden Behandlungsschemata ab Woche 6 zugeteilt: Entyvio 300 mg alle acht Wochen, Entyvio 300 mg alle vier Wochen oder Placebo alle vier Wochen. Placebo-Patienten aus der Induktionsphase wurden bis Woche 52 mit Placebo weitergeführt. Bei Patienten, bei denen in Woche 6 ein klinisches Ansprechen nachweisbar war, wurde ein Kortikosteroid-Ausschleichschema begonnen. Der primäre Endpunkt war der Anteil der Patienten in klinischer Remission nach Woche 52. In Tabelle 4 sind die primären und sekundären Endpunkte der Erhaltungsphase zusammengefasst. Entyvio war bezüglich klinischer Remission sowohl bei TNFα-Antagonisten-naiven Patienten als auch bei Patienten mit einer erfolglosen TNFα-Antagonisten-Vortherapie gegenüber Placebo überlegen. Explorative Analysen lieferten zusätzliche Daten zu wichtigen Teilpopulationen der Studie. Etwa bei 50% der Patienten von GEMINI II hatte zuvor die Behandlung mit TNFα-Antagonisten versagt. Von diesen Patienten zeigten 28% der mit Entyvio alle acht Wochen und 27% der mit Entyvio alle vier Wochen behandelten Patienten sowie 13% der Patienten unter Placebo nach Woche 52 eine klinische Remission (95%-Konfidenzintervall für die Differenz zu Placebo 3–28% bzw. 2-27%). Eine Kortikosteroid-freie Remission war bei 24%, 16% und 0% der Patienten, die Entyvio alle 8 Wochen, alle 4 Wochen und Placebo erhielten, zu verzeichnen. Bei Patienten mit komedizierten Immunmodulatoren (Azathioprin, 6-Mercaptopurin) zu Studienbeginn (Woche 0) zeigten in Woche 52 46% unter Entyvio alle acht Wochen und 47% unter Entyvio alle vier Wochen sowie 31% unter Placebo eine klinische Remission (jeweilige 95-%-Konfidenzintervalle für die Differenz zur klinischen Remissionsrate der Placebogruppe-4%–34% bzw. -2%–35%). Von den Patienten mit komedizierten Kortikosteroiden zu Studienbeginn (Woche 0) zeigten in Woche 52 39% unter Entyvio alle acht Wochen und 35% unter Entyvio alle vier Wochen sowie 17% unter Placebo eine klinische Remission (jeweilige 95%-Konfidenzintervalle für die Differenz zur klinischen Remissionsrate der Placebogruppe 9–35% bzw. 5–31%). Unter Entyvio wurden bis Woche 52 klinisch bedeutsame Verbesserungen festgestellt, die signifikant stärker ausgeprägt waren als in der Placebogruppe. Dies betraf den EQ-5D- und EQ-5D-VAS-Score, den IBDQ-Gesamtscore und die IBDQ-Subskalen der Darmsymptome und systemische Funktion. Die Daten der laufenden offenen Extensionsstudie GEMINI OLE mit Entyvio alle 4 Wochen sind mit einem anhaltenden Nutzen hinsichtlich klinischer Remission und klinischem Ansprechen bis zu 124 Wochen vereinbar. Diese offene Extensionsstudie schloss 726 Patienten mit M. Crohn aus GEMINI II ein. Verzögertes Ansprechen: Patienten in GEMINI II, die nach Woche 6 kein Ansprechen zeigten, verblieben in der Studie und erhielten alle vier Wochen Entyvio. Ein verbessertes klinisches Ansprechen lag nach Woche 10 und Woche 14 bei einem grösseren Anteil der Entyvio-Patienten (16% bzw. 22%) vor als in der Placebogruppe (7% bzw. 12%). Wiederaufnahme der Therapie: Patienten, die unter Entyvio in Woche 6 angesprochen hatten und dann nach Placebo re-randomisiert wurden und im weiteren Verlauf einen Verlust des Ansprechens erfuhren, durften in die laufende offene Extensionsstudie GEMINI OLE wechseln, wo sie Entyvio alle vier Wochen erhielten. Bei 46% dieser Patienten wurde eine klinische Remission nach Woche 28 erreicht, bei 41% nach Woche 52. Dosiseskalation: Patienten in GEMINI II, die nach anfänglichem Ansprechen mit Entyvio alle acht Wochen einen Verlust des Ansprechens erfuhren, konnten in die offene Extensionsstudie GEMINI OLE wechseln, wo sie Entyvio alle vier Wochen erhielten. Bei 23% dieser Patienten war eine klinische Remission in Woche 28 und bei 32% in Woche 52 zu verzeichnen. Hospitalisationen, operative Eingriffe, Darmresektionen: In GEMINI II betrug bis Woche 52 der Anteil der CD-bedingten Krankenhausaufenthalte, operative Eingriffe und Darmresektionen unter TNFα-Antagonisten-naiven Patienten und Patienten nach erfolgloser TNFα-Antagonisten-Vortherapie mit Entyvio alle 8 Wochen, alle vier Wochen oder Placebo 12.1%, 12.7% und 12.07% bzw. 11.0%, 5.2% und 11.5%. In der laufenden offenen Extensionsstudie GEMINI OLE, welche allen Entyvio-Patienten ungeachtet ihres Status in der Studie GEMINI II offen stand, betrug die Kaplan-Meier-Schätzung für den Anteil CD-bedingter Krankenhausaufenthalte, operativer Eingriffe und Darmresektionen unter TNFα-Antagonisten-naiven Patienten und Patienten mit erfolgloser TNFα-Antagonisten-Vortherapie 12.4% und 18.4% nach 1 Jahr sowie 18.2% und 23.4% nach 2 Jahren. Unter den total 700 aus GEMINI II aufgenommenen Patienten hatte bei 53% eine Therapie mit einem TNFα-Antagonisten zuvor versagt und 39% von denen, die in der Induktionsphase Entyvio erhielten, hatten bis Woche 6 kein klinisches Ansprechen gezeigt. Unter den total 372 aus GEMINI III aufgenommenen Patienten hatte bei 75% eine Therapie mit einem TNFα-Antagonisten zuvor versagt und 45% von denen, die in der Induktionsphase Entyvio erhielten, hatten bis Woche 6 kein klinisches Ansprechen gezeigt. Weitere Informationen Die Studie GEMINI III (n = 416) war eine weitere randomisierte, doppelblinde, placebokontrollierte Studie zur Beurteilung der Wirksamkeit nach Woche 6 und Woche 10 bei Patienten mit erfolgloser TNFα-Antagonisten Vortherapie. Die Studienpopulation war identisch zu GEMINI II, ausser dass 75% Patienten mit erfolgloser TNFα-Antagonisten -Vortherapie eingeschlossen waren. Die Patienten wurden mittels doppelblinder Randomisierung (1:1) der Behandlung mit Entyvio 300 mg oder Placebo in Woche 0, 2 und 6 zugeteilt. Der primäre Endpunkt war der Anteil der Patienten in klinischer Remission nach Woche 6 in der Subpopulation derjenigen Patienten, bei denen zuvor TNFα-Antagonisten versagt hatten. Dieser primäre Endpunkt wurde bei 15% der Patienten, die Entyvio erhielten und bei 12% der Patienten unter Placebo erreicht und erlangte keine statistische Signifikanz. Daher können nachfolgende Analysen der sekundären Endpunkte nur als explorativ angesehen werden. In der Subpopulation mit erfolgloser Vorbehandlung mit TNFα-Antagonisten der GEMINI III Studie wurde nach zehnwöchiger Behandlung mit Entyvio bei 27% der Patienten eine klinische Remission erreicht, verglichen mit 12% der Patienten, die Placebo erhielten; ein verbessertes klinisches Ansprechen in Woche 6 bzw. 10 wurde bei 39% bzw. 47% der Patienten unter Entyvio und 22% bzw. 25% der Patienten unter Placebo erzielt. In der Gesamt-Studienpopulation wurde nach sechs- bzw. zehnwöchiger Behandlung mit Entyvio bei 19% bzw. 29% der Patienten eine klinische Remission erreicht, verglichen mit 12% und 13% der Patienten, die Placebo erhielten; ein verbessertes klinisches Ansprechen wurde bei 39% bzw. 48% der Patienten unter Entyvio und 23% bzw. 24% der Patienten unter Placebo erzielt. Extensionsstudie In der laufenden offenen Extensionsstudie GEMINI OLE wurden 675 Patienten aus GEMINI I, 726 Patienten aus GEMINI II und 384 Patienten aus GEMINI III aufgenommen. Interimsresultate sind unter den entsprechenden Abschnitten zu Colitis ulcerosa und M. Crohn beschrieben. Pharmakokinetik Die Pharmakokinetik von Vedolizumab nach ein- und mehrmaliger Anwendung ist bei gesunden Probanden und bei Patienten mit mittel- bis hochgradig aktiver Colitis-ulcerosa- oder Morbus-Crohn-Erkrankung untersucht worden. Das pharmakokinetische Profil von Vedolizumab und die Auswirkungen verschiedener Kovariablen wurden in einem populationskinetischen Modell charakterisiert. Absorption Bei den Patientenpopulationen mit Colitis ulcerosa und mit Morbus Crohn wurden vergleichbare pharmakokinetische Eigenschaften beobachtet. Vedolizumab zeigt bei Serumkonzentrationen über 1 mcg/ml eine lineare Pharmakokinetik. Bei den Patienten, denen in Woche 0 und 2 je 300 mg Vedolizumab als 30-minütige intravenöse Infusion verabreicht wurden, betrug der mittlere Serum-Talspiegel nach Woche 6 bei Colitis ulcerosa 27.9 mcg/ml (SD ± 15.51) und bei Morbus Crohn 26.8 mcg/ml (SD ± 17.45). Ab Woche 6 erhielten die Patienten 300 mg Vedolizumab alle acht oder vier Wochen. Bei den Patienten mit Colitis ulcerosa betrug der mittlere Serum-Talspiegel im Gleichgewichtszustand 11.2mcg/ml (SD±7.24) bzw. 38.3 mcg/ml (SD ± 24.3). Bei den Patienten mit Morbus Crohn betrug der mittlere Serum-Talspiegel im Gleichgewichtszustand 13.0mcg/ml (SD±9.08) bzw. 34.8 mcg/ml (SD ± 22.55). Distribution Das Verteilungsvolumen von Vedolizumab wurde auf rund 5 Liter geschätzt. Die Plasmaproteinbindung von Vedolizumab wurde nicht untersucht. Eine 450 mg Entyvio-Dosis war nach intravenöser Gabe im Liquor cerebrospinalis gesunder Probanden nicht nachzuweisen. Elimination Die Clearance wurde auf 0.157 l/Tag und die Plasma-Halbwertszeit auf ungefähr 25 Tage geschätzt. Der genaue Eliminationsmechanismus ist aber nicht bekannt. Die Clearance schien bei höherem Körpergewicht und Vorliegen von Anti-Vedolizumab-Antikörpern erhöht zu sein. Anwendung bei besonderen Patientengruppen Im populationskinetischen Modell war kein Einfluss des Alters auf die Clearance festzustellen. Es wurden keine formalen Studien zu den Auswirkungen einer eingeschränkten Nieren- oder Leberfunktion auf die Pharmakokinetik von Vedolizumab durchgeführt. Präklinische Daten Die spezifische Hemmung des α4β7/MAdCAM-Signalwegs führt in vivo zu einer selektiven Wirkung im Darm. Sie lindert bei Affen die gastrointestinale Entzündung, ohne die Immunantwort auf dermale Antigen-Provokation oder die Immunüberwachung des ZNS zu beeinflussen. Basierend auf den konventionellen Studien zur Sicherheitspharmakologie wird von keinen besonderen Gefahren für den Menschen ausgegangen. In Toxizitätsstudien resultierte die wiederholte Vedolizumabgabe in lymphoider Depletion der Peyer'schen Plaques. Ein Bezug der in den Toxizitätsstudien bei Affen beobachteten, nicht vollständig reversiblen, lymphoplasmozytischen Gastritis mit gesteigerter Regeneration der Mucosa zur wiederholten Vedolizumabgabe kann nicht ausgeschlossen werden. Die Relevanz der bei den höchsten verwendeten Dosen isoliert bei 2 Kaninchen und einem Affen beobachteten Gliosen für den Menschen ist als gering eingestuft worden. Langfristige tierexperimentelle Studien zu Vedolizumab zur Untersuchung seines karzinogenen Potenzials wurden nicht durchgeführt, da kein pharmakologisch relevantes Modell existiert. Bei einer pharmakologisch responsiven Spezies (Langschwanzmakaken) ergaben 13- und 26-wöchige Studien zur Toxikologie keine Hinweise auf zelluläre Hyperplasie oder systemische Immunmodulation, die potenziell mit Onkogenese assoziiert sein könnte. In vitro wurden ausserdem keine Auswirkungen von Vedolizumab auf die Proliferationsrate oder Zytotoxizität einer humanen Tumorzelllinie mit Expression von α4β7-Integrin festgestellt. Es wurden keine dedizierten präklinischen Fertilitätsstudien zu Vedolizumab durchgeführt. Nach Verabreichung von Vedolizumab an trächtige Langschwanzmakaken während des Grossteils der Tragezeit (insbesondere zwischen dem 20. und 140. Tag; eine durchschnittliche Tragezeit beträgt 160 Tage) waren keine Hinweise auf Auswirkungen auf Teratogenität oder prä- und postnatale Entwicklung bei Jungtieren bis zu 6 Monaten zu erkennen. Die bei Dosen ≥30 mg/kg Q2W beobachteten skelettalen Variationen (Rippenvergrösserung, Zusatzrippen, Brustbeinfehlstellung) wurden, da innerhalb der historischen Kontrollen liegend, als toxikologisch nicht relevant erachtet. In geringer Konzentration (<300 Mikrogramm/l) war Vedolizumab an Tag 28 post partum in der Milch von Langschwanzmakaken nachzuweisen, denen 100 mg/kg Vedolizumab alle 2 Wochen gegeben worden waren. Nicht aber bei Tieren, die 10 mg/kg erhielten. Ein Bezug der in einem Nachkommen eines Hochdosismuttertieres (100 mg/kg Q2W), welches vorzeitig aus der Studie genommen werden musste, beobachteten Atrophie der intestinalen Mucosa und lymphoiden Atrophie der Peyer'schen Plaques zur wiederholten Vedolizumabgabe ist anzunehmen. Der auf der systemischen Exposition (AUC) am NOAEL von 30 mg/kg Q2W basierende Sicherheitsabstand beträgt das 18-Fache des Sicherheitsabstands basierend auf Toxizitätsstudien bei Affen. Sonstige Hinweise Inkompatibilitäten Da keine Inkompatibilitätsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden. Haltbarkeit Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden. Die rekonstituierte bzw. verdünnte Lösung enthält keine Konservierungsmittel. Die chemische und physikalische Stabilität der rekonstituierten bzw. verdünnten Lösung ist für 12 Stunden bei Raumtemperatur (15–25 °C) und für 24 Stunden bei 2–8 °C nachgewiesen. Aus mikrobiologischen Gründen sollte die gebrauchsfertige Zubereitung möglichst sofort verwendet werden. Wenn die gebrauchsfertige Zubereitung nicht sofort verwendet wird, liegen Aufbrauchfristen und Lagerbedingungen in der Verantwortung des Anwenders, dürfen jedoch insgesamt 24 Stunden nicht überschreiten. Die kumulative Lagerungszeit der rekonstituierten Lösung und der verdünnten Lösung (Infusionslösung) darf nicht 12 Stunden bei Raumtemperatur (15-25 °C) oder 24 Stunden bei 2-8 °C überschreiten. Rekonstituierte oder verdünnte Lösung nicht einfrieren. Besondere Lagerungshinweise Für Kinder unzugänglich aufbewahren. Im Kühlschrank lagern (2–8 °C). Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen. Zu den Lagerungsbedingungen nach der Rekonstitution des Arzneimittels siehe Rubrik «Haltbarkeit». Hinweise für die Handhabung Jede Durchstechflasche ist nur zum einmaligen Gebrauch bestimmt. Rekonstitution Entyvio sollte bei 20-25 °C rekonstituiert werden. Bei der Zubereitung der Entyvio-Lösung zur intravenösen Infusion aseptisch arbeiten. Schnappdeckel von der Durchstechflasche entfernen und Flasche mit Alkoholtupfer abwischen. Mit einer Spritze mit 21- bis 25-Gauge-Nadel Vedolizumab mit 4.8 ml sterilem Wasser für Injektionszwecke rekonstituieren. Nadel mittig durch den Stopfen in die Flasche stechen und Flüssigkeitsstrom an die Wand des Gefässes lenken, um übermässiges Schäumen zu vermeiden. Durchstechflasche mindestens 15 Sekunden behutsam schwenken. Nicht kraftvoll schütteln oder über Kopf kippen. Durchstechflasche bis zu 20 Minuten bei 20-25 °C ruhen lassen, sodass die Rekonstitution ablaufen und eventueller Schaum sich absetzen kann; in dieser Zeit kann die Flasche geschwenkt und der Lösungsfortschritt überprüft werden. Wenn die Auflösung nach 20 Minuten nicht vollständig abgeschlossen ist, weitere 10 Minuten warten. Die rekonstituierte Lösung vor der Verabreichung visuell auf Partikel und Verfärbung inspizieren. Die Lösung soll klar oder opalisierend, farblos bis hellgelb und frei von sichtbaren Partikeln sein. Rekonstituierte Lösung mit untypischer Farbe oder mit Partikeln darf nicht verabreicht werden: Wenn diese rekonstituierte Lösung der Zulassungsinhaberin zwecks Untersuchung nicht zurückgegeben werden kann, soll sie verworfen werden. Herstellung der Infusionslösung Vor der Entnahme der rekonstituierten Lösung aus der Durchstechflasche die Flasche 3-mal behutsam über Kopf kippen. 5 ml (300 mg) rekonstituiertes Entyvio mit einer Spritze mit 21- bis 25-Gauge-Nadel entnehmen. Die 5 ml (300 mg) rekonstituiertes Entyvio zu 250 ml steriler 0.9-%iger Natriumchloridlösung zugeben und Infusionsbeutel vorsichtig mischen (es ist nicht notwendig, vor der Zugabe des Entyvio 5 ml der 0.9-%igen Natriumchloridlösung aus dem Infusionsbeutel zu entnehmen). Der gebrauchsfertigen Infusionslösung bzw. dem intravenösen Infusionsbesteck keine anderen Arzneimittel hinzufügen. Infusionslösung über einen Zeitraum verabreichen, der den empfohlenen Zeitraum nicht unterschreitet (siehe Rubrik «Dosierung/Anwendung»). Nicht verbrauchte Reste der Infusionslösung verwerfen. Entsorgung Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu entsorgen. Zulassungsnummer 63285 (Swissmedic). Zulassungsinhaberin Takeda Pharma AG, Freienbach 。用药温馨提示:当您服用此药物时,需定期接受医疗专业人士的检查,以便随时针对其药效、副作用等情况进行监测。本网站所包含的信息旨在为患者提供帮助,不能代替医学建议和治疗。
药品价格查询,专业药品查询网站,药品说明书查询,药品比价 » 维多珠单抗冻干粉注射剂(vedolizumab)-维多珠单抗中英文说明书-Entyvio Trockensubstanz 300mg
药品价格查询,专业药品查询网站,药品说明书查询,药品比价 » 维多珠单抗冻干粉注射剂(vedolizumab)-维多珠单抗中英文说明书-Entyvio Trockensubstanz 300mg